Secreted a disintegrin-like and metalloprotease with thrombospondin type 1 motif (ADAMTS) proteases play crucial roles in tissue development and homeostasis. The biological and pathological functions of ADAMTS proteases are determined broadly by their respective substrates and their interactions with proteins in the pericellular and extracellular matrix. For some ADAMTS proteases, substrates have been identified and substrate cleavage has been implicated in tissue development and in disease. For other ADAMTS proteases, substrates were discovered in vitro, but the role of these proteases and the consequences of substrate cleavage in vivo remains to be established. Mutations in ADAMTS10 and ADAMTS17 cause Weill–Marchesani syndrome (WMS), a congenital syndromic disorder that affects the musculoskeletal system (short stature, pseudomuscular build, tight skin), the eyes (lens dislocation), and the heart (heart valve abnormalities). WMS can also be caused by mutations in fibrillin-1 (FBN1), which suggests that ADAMTS10 and ADAMTS17 cooperate with fibrillin-1 in a common biological pathway during tissue development and homeostasis.
1. The ADAMTS Protease Family
The family of
a disintegrin-like
and
metalloprotease with
thrombo
spondin type 1 motif (ADAMTS) comprises 19 secreted metalloproteases, which are primarily involved in the formation, remodeling and/or degradation of components of the extracellular matrix (ECM)
[1]. All of the ADAMTS proteases share a similar domain organization with a conserved N-terminal protease domain and a variable C-terminal ancillary domain, which is implicated in substrate recognition and ECM binding (a)
[2][3][4]. The conserved ADAMTS protease domain consists of a signal peptide to target ADAMTS proteases for secretion, a propeptide that is typically removed by furin/PACE proprotein convertases to activate ADAMTS proteases, the catalytic metalloproteinase domain itself, and a disintegrin-like domain. The ancillary domain, which mediates binding to ECM components and specific protease substrates, consists of a conserved central thrombospondin type 1 sequence repeat motif (TSR), a cysteine-rich domain and a spacer domain. The subsequent C-terminal domains and their arrangement distinguish individual ADAMTS proteases and can comprise combinations of up to 14 TSRs, CUB domains, a mucin/proteoglycan domain, or the GON-1 domain
[5].
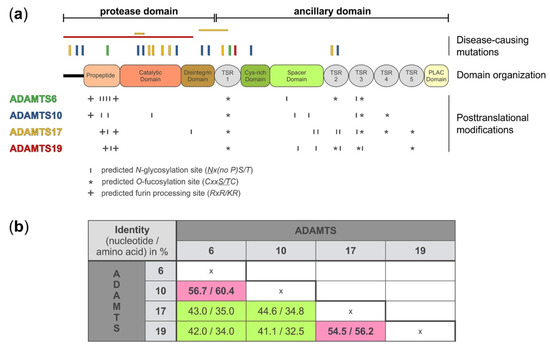
Figure 1. A disintegrin-like and metalloprotease with thrombospondin type 1 motif (ADAMTS)6, 10, 17, and 19 domain organization, location of disease-causing mutations, and sites for putative post-translational modifications. (
a) Domain organization, depicting the protease and ancillary domain of ADAMTS6, 10, 17, and 19, which have an identical domain organization. However, the four ADAMTS proteases show differences in the number and localization of predicted sites for posttranslational modifications, such as N-glycosylation (
http://www.cbs.dtu.dk/services/NetNGlyc/), O-fucosylation, and furin processing which could specify individual substrates, define protein-protein interactions, or govern autocatalytic properties that collectively distinguish these four ADAMTS proteases from each other. The location of disease-causing mutations is indicated on top of the diagram. Point mutations are indicated with vertical bars and deletion of larger gene fragments are indicated with horizontal bars
[6][7][8][9][10][11][12][13][14]. The disease-causing mutations depicted for each ADAMTS protease are color-coded to match the color of the individual ADAMTS protease in panel a (left). (
b) Nucleotide and amino acid sequence identity for ADAMTS6, 10, 17, and 19 shows higher sequence identity for ADAMTS6 and ADAMTS10, and ADAMTS17 and ADAMTS19, respectively (pink). The following DNA/protein sequences from the NCBI database were used to calculate the percentage identity after sequence alignment with Clustal Omega (
https://www.ebi.ac.uk/Tools/msa/clustalo/): ADAMTS6: NM_197941.4/NP_922932.2; ADAMTS10 NM_030957.4/NP_112219.4; ADAMTS17 XM_005254872.3/XP_005254929.1; ADAMTS19 NM_133638.6/NP_598377.3.
The individual proteases of the ADAMTS family can be grouped according to their evolutionary homology and their biological substrates, e.g., proteoglycanases (ADAMTS1, 4, 5, 8, 9, 15, and 20) or procollagen N-propeptidases (ADAMTS2, 3, and 14). Until about a decade ago, no substrates were reported for the ADAMTS proteases that form the central clade of the ADAMTS homology tree, i.e., ADAMTS6, 7, 10, 12, 16, 17, 18, and 19, and these ADAMTS proteases were considered “orphan”
[15][16][17]. However, more recent work identified several substrates for these proteases, many of them are linked to fibrillin biology, such as fibrillin-1 and fibrillin-2 themselves, several latent transforming growth factor (TGF) β binding proteins (LTBPs), fibronectin, or ADAMTS-like 6
[18][19][20][21][22]. The consilience between evolutionary conservation and functionally related ECM substrates suggests that at least some of these ADAMTS proteases perform biological roles related to the formation or function of fibrillin microfibrils
[23]. Fibrillin microfibrils represent pivotal ECM signaling platforms integrating TGFβ, bone morphogenetic protein (BMP), and mechanosignaling
[24]. The fact that mutations in
ADAMTS10,
ADAMTS17, fibrillin-1 (
FBN1),
LTBP2, or
LTBP3 can cause almost identical short stature syndromes, called acromelic dysplasias, further supports the concept that some ADAMTS proteases in this central clade, specifically ADAMTS10 and ADAMTS17, are functionally connected through their ECM substrates
[23][6][25][26]. Here, we review the recent literature on the homologous protease pairs ADAMTS6/ADAMTS10 and ADAMTS17/ADAMTS19. We explore the connection of ADAMTS10 and ADAMTS17 to fibrillin microfibril biology based on the consilience in human genetic disorders, sequence homology, and experimental evidence, and we develop a conceptual model of how these proteases may interact and cooperate in the pericellular matrix (PCM) and the ECM. We also include a discussion of the respective sister proteases, ADAMTS6 and ADAMTS19, since it is known for other ADAMTS protease pairs that they can cooperate or functionally compensate for each other during tissue development or in tissue homeostasis
[27][28][29].
2. Domain Organization and Posttranslational Modifications of ADAMTS6, 10, 17, and 19
On the protein level, ADAMTS6, 10, 17, and 19 share the same domain organization (a). However, each protease pair arose from distinct gene duplication events
[30]. When comparing the nucleotide and amino acid sequences between the four proteases, it is evident that ADAMTS10 sequences are more similar to ADAMTS6, where 60% of the amino acid residues are identical, and ADAMTS17 sequences are more similar to ADAMTS19, with 56% of the amino acid residues being identical (b)
[1][31]. Despite the evolutionary homology, the identical domain organization, and the relatively high amino acid sequence identity, the ADAMTS proteases that form the individual protease pairs, ADAMTS6/ADAMTS10 and ADAMTS17/ADAMTS19, appear to have distinct biological functions, based on their involvement in different human disorders (see below). One possible explanation for the diversification in the function of these proteases could be differences in posttranscriptional and posttranslational modifications.
Alternative splicing is a posttranscriptional mechanism that can expand the diversity and thus function of ADAMTS proteases by generating different isoforms. ADAMTS proteases have several splice variants based on the NCBI protein database. There are 13 isoforms listed for ADAMTS6, 4 for ADAMTS10, 12 for ADAMTS17, and 5 for ADAMTS19. For most of these isoforms, tissue-specific expression or functional data are not available. However, by homology mapping with ADAMTS10 as a template and analysis of expressed sequence tags in the GenBank™ database the existence of at least two splice variants for ADAMTS6 were predicted and subsequently shown experimentally in epithelial cells
[32][33]. In addition, northern blot analysis of total mRNA isolated from adult human tissue demonstrated two ADAMTS10 mRNA species that differed in size, suggesting alternative splicing of ADAMTS10 mRNA as well
[33]. Two isoforms of ADAMTS17 with distinct expression patterns have been described previously
[10]. Our own unpublished data show expression of at least three additional isoforms of ADAMTS17 that differ in the sequence of the spacer domain (Balic, et al., manuscript in preparation).
In addition to alternative splicing, ADAMTS6, 10, 17, and 19 show differences in the number and location of predicted and experimentally verified posttranslational modifications, such as furin/PACE-processing, autocatalysis, N-glycosylation, or O-fucosylation. Based on western blot analysis, ADAMTS6 and ADAMTS19 are furin-processed but do not undergo apparent autocatalysis (Karoulias et al., unpublished data for ADAMTS19)
[34]. However, a direct comparison between active ADAMTS6 and an inactive mutant form was not shown to completely rule out the possibility of ADAMTS6 autocatalysis. The propeptide of ADAMTS17 is also processed by furin, but in contrast to ADAMTS6 and ADAMTS19, ADAMTS17 undergoes extensive autoproteolysis at the cell surface or in the ECM
[22]. Furin-processing of the ADAMTS17 propeptide is not required to activate ADAMTS17. Instead, the ADAMTS17 propeptide may act as a chaperone to facilitate ADAMTS17 secretion, since removal of the propeptide did abolish ADAMTS17 secretion or its release from the cell surface
[22]. A similar role was previously described for the propeptide of ADAMTS9
[35]. ADAMTS10 on the other hand has a degenerated consensus sequence for furin-processing (GLKR instead of RLKR) and thus the propeptide remains covalently associated with the ADAMTS10 protease after secretion. Wild type ADAMTS10 has weak protease activity against fibrillin-1
[36]. However, upon restoration of the consensus furin-processing site in recombinant ADAMTS10, the propeptide was efficiently excised and proteolytic activity of ADAMTS10 against fibrillin-1 was enhanced
[36].
ADAMTS proteases can be N-glycosylated and O-fucosylated. ADAMTS6 and ADAMTS10 contain six predicted N-glycosylation sites but their location is different (a and ). For example, four of the six predicted N-glycosylation sites in ADAMTS6 are located in the propeptide domain while ADAMTS10 harbors only two of the six predicted N-glycosylation sites in the propeptide. Interestingly, one of the predicted N-glycosylation sites in ADAMTS10 is located in the catalytic domain and it is tempting to speculate that this site may modulate proteolytic activity if differentially glycosylated. ADAMTS17 has seven predicted N-glycosylation sites with five of them being located in the ancillary domain. In contrast, ADAMTS19 contains only five predicted N-glycosylation sites. In addition to N-glycosylation, TSR domains of ADAMTS proteases can undergo O-fucosylation at the Cxx(S/T)C consensus motif as part of a non-canonical quality control pathway in the endoplasmic reticulum
[37][38]. Three of the five TSR domains of ADAMTS6 and ADAMTS10 and four of the five TSR domains of ADAMTS17 and ADAMTS19 contain the consensus sequence for O-fucosylation
[39]. For ADAMTS17, it was shown that TSR1, 3, and 5 were fully modified
[22]. TSR4 was not O-fucosylated, despite the presence of the consensus motif. Functionally, the absence of O-fucosylation on TSR3 and TSR5, and to a lesser extent on TSR1, resulted in the lack of secretion of recombinant ADAMTS17
[22]. It is anticipated that O-fucosylation of TSRs in ADAMTS6, 10, and 19 would play a similar role in protein quality control and protein secretion. However, experimental data is lacking and the degree of O-fucosylation needs to be determined for each of these proteases.
Table 1. Comparison of features distinguishing the individual ADAMTS6, 10, 17, and 19 proteases.
| |
ADAMTS6 |
ADAMTS10 |
ADAMTS17 |
ADAMTS19 |
| N-Glycosylation sites 1 |
6 |
6 |
7 |
5 |
| O-Fucosylation sites 2 |
3 |
3 |
4 |
4 |
| Furin consensus sequences 3 |
2 |
1 |
2 |
2 |
| Furin processing 4 |
yes |
no |
yes |
yes |
| Autocatalysis |
no |
no |
yes |
no |
| Substrates |
LTBP1, SDC4 |
FBN1, FBN2 |
ADAMTS17 |
n.d. |
| Alternative splicing |
yes |
yes |
yes |
likely |
| Human disorders |
Prolonged QRS syndrome |
WMS 1
(MIM #277600) |
WMS 4
(MIM #613195) |
Non-syndromic heart valve disease |
| Disease-causing human mutations |
2 |
9 |
9 |
2 |
| Gene knockout phenotype in mice |
Prenatal/neo-natal lethality, double outlet right ventricle, ventricular hypertrophy,
atrial and ventricular septal defects |
Some prenatal/ neonatal lethality, abnormal ciliary zonule, shorter long bones due to growth plate abnormalities, skeletal muscle abnormalities |
Some prenatal/ neonatal lethality, skeletal growth impairment due to growth plate abnormalities, brachydactyly by 8 mos. of age |
Aortic valve dysfunction in ~40% of knockout mice |
| Fibrillin-1 binding |
n.d. |
yes |
yes |
n.d. |
| Fibrillin microfibril formation 5 |
n.d. |
Promotes FBN1 deposition |
No effect |
n.d. |
Taken together, different isoforms of ADAMTS6/ADAMTS10 and ADAMTS17/ADAMTS19 proteases, generated by alternative splicing or differential posttranslational modifications, could be important determinants for the regulation of ADAMTS secretion or cell surface localization, for ADAMTS activation or modulation of protease activity, or for substrate recognition and ECM binding, and thus may provide the molecular underpinnings for differences in their biological functions.
3. Gene Expression Patterns of ADAMTS6, 10, 17, and 19
The biological functions of ADAMTS6, 10, 17, and 19 are not only determined by differential posttranscriptional and posttranslational modifications, which may guide ECM localization and substrate binding and cleavage, but also by the cell types and tissues that express these proteases.
Adamts10 for example is widely expressed in most adult tissues in mice and almost universally expressed during mouse embryonic development with the exception of the ectoderm
[20][33]. RNA in situ hybridization demonstrated dynamic
Adamts10 expression, which was generally low during early embryonic development [embryonic day (E) 9.5 to E12.5] and induced in later stages of development (E14.5 and E17.5)
[33]. At E14.5,
Adamts10 was expressed in several craniofacial tissues, notably in the perichondrium and periosteum, but not in cartilage, of newly forming bones of the mandible, and in the tongue muscle. In developing lungs,
Adamts10 was expressed in the connective tissue between the bronchi and the accompanying blood vessels.
Adamts10 was also expressed in the stomach, duodenum, pancreas, dorsal root ganglia, and primary ossification centers of vertebrae, but was absent in the liver. In the developing musculoskeletal system,
Adamts10 mRNA was detected between the cartilaginous metacarpals and metatarsals of the hand and feet and in dense connective tissues such as the joint capsule and developing tendons and ligaments. At E17.5
Adamts10 expression was strong in the cartilage of developing bones and in the wall of large arteries.
Adamts10 was expressed in several ocular tissues during development, including non-pigmented ciliary epithelium, lens fiber cells, and parts of the retina
[20]. Expression of ADAMTS10 in the eye is relevant for the characteristic ectopia lentis phenotype observed in individuals with Weill–Marchesani syndrome (WMS). All these data support a role for ADAMTS10 in the early steps of tissue and organ formation.
Adamts10 continued to be expressed in adult chondrocytes, tendon, and skeletal muscle in mice
[20]. In human adult tissue,
ADAMTS10 mRNA was expressed in the heart, brain, lung, pancreas, but
ADAMTS10 expression was notably low or absent in skeletal muscle
[33]. Given the broad expression of ADAMTS10 during embryonic development, it was somewhat surprising, that most
Adamts10 knockout mice apparently undergo normal development, with limited embryonic lethality (see below)
[19][20].
Similar to
ADAMTS10,
ADAMTS17 is widely expressed in many fetal and adult tissues in humans
[10]. Two isoforms of
ADAMTS17, isoform a (22 exons) and isoform b (16 exons), with distinct expression patterns, were identified. Both
ADAMTS17 isoforms were expressed in lung, brain, and the eye, including the retina. Immunostaining in human normal and cancer tissues showed widespread expression in multiple tissues to varying degrees
[10]. However, no images were shown and the reported localization of ADAMTS17 in the cytoplasm raises the possibility that the antibody used in this study was non-specific in tissue staining, even though it recognizes recombinant ADAMTS17 protein
[22]. In-situ hybridization studies in mouse embryonic tissue at E16.5 and in neonates demonstrated
Adamts17 expression in the eye, especially at the lens equator and in the trabecular region
[22]. Apart from the eye,
Adamts17 mRNA was detected in the perichondrium, the intervertebral disc, and in the smooth muscle cell layer of blood vessel walls. The most pronounced expression of
Adamts17 was observed in lung parenchyma, skin epidermal basal cell layer, and in developing hair follicles.
Adamts17 mRNA was notably absent from the growth plate of long bones, the endothelium of blood vessels, and the bronchial epithelium in lung tissue.
Less detailed information about the expression patterns of
ADAMTS6 and
ADAMTS19 is currently available.
ADAMTS6 mRNA was detected in the mouse heart, specifically in the outflow tract, the heart valves, the atria, and the ventricular myocardium
[34]. In northern blot analysis,
ADAMTS6 mRNA was weakly expressed in placental tissue, but was not detected in mouse embryonic development or in adult human tissues
[40].
ADAMTS19 mRNA was detected in fetal lung and osteosarcoma tissue
[41]. More recently,
Adamts19 was shown to be expressed in the heart valve at all stages of heart valve formation and elongation and in bone and cartilage anlagen at E10.5
[42].
Taken together,
ADAMTS10 and
ADAMTS17 show broad, both overlapping and distinct gene expression patterns in tissues. Therefore, it is possible that ADAMTS10 and ADAMTS17 may compensate for each other in mouse knockout models of the individual proteases at least in some tissues (see below). If these proteases can indeed compensate for each other in murine loss-of-function models, similar to what has been observed for the ADAMTS7/ADAMTS12 and ADAMTS9/ADAMTS20 pairs, needs to be explored
[27][28]. No compensation of
ADAMTS10 with
ADAMTS17 or vice versa appears to occur in tissues affected in WMS, specifically the eye and the musculoskeletal system. However, a lack of cardiac involvement and joint contractures in WMS4 due to
ADAMTS17 mutations leaves the possibility that compensation with
ADAMTS10, but not vice versa, causes the lack of cardiac and joint presentations in WMS4 (see below).
ADAMTS6 and
ADAMTS19 appear to have more tissue-specific expression patterns, which are reflected in the specific diseases associated with mutations in
ADAMTS6 and
ADAMTS19, as described in the next section.
 +1 point
+1 point