1000/1000
Hot
Most Recent
 +1 point
+1 point
As we age, our bodies accrue damage in the form of DNA mutations. These mutations lead to the generation of sub-optimal proteins, resulting in inadequate cellular homeostasis and senescence. The build-up of senescent cells negatively affects the local cellular micro-environment and drives ageing associated disease, including neurodegeneration. Therefore, limiting the accumulation of DNA damage is essential for healthy neuronal populations.
An exposure to low oxygen conditions (hypoxia) perturbs oxidative phosphorylation, which in turn leads to an increase in cellular concentration of reactive oxygen species (ROS). ROS can cause oxidative stress, which is capable of ageing human cells through mutation induction and protein oxidation, potentially causing senescence and neurodegeneration [1][2][3][4]. Moreover, Naked Mole Rats (NMR) display a high prevalence of protein plaques (tau and amyloid β), which are currently believed to be a significant cause of neurodegeneration, but fail to exhibit any signs of neuronal death [5]. This review aims to summarise current knowledge on molecular pathways that NMRs employ to ensure genome integrity and minimal cellular senescence to achieve neuroprotection.
Specifically, upon DNA damage induction, both NMR and human cells increased transcription of repair factors associated with non-homologous end joining (NHEJ), mismatch repair (MMR), homology-directed repair (HDR) and base excision repair (BER) [6]. Higher transcription rates of DDR genes triggered by DNA damage might ensure a large mRNA pool for DDR protein synthesis and subsequent DNA repair. As a result, NMR BER resolved DNA damage more rapidly, showed improved efficiency of excision and faster return to basal transcription levels, when compared to the mouse [6][7][8]. In humans and NMRs, increased DDR gene transcription and efficient DNA repair correlates with a longer life span [6].
Microhomology-mediated end joining (MMEJ) is an error-prone repair pathway, which requires both PARP1 and XRCC1 and is capable of causing large genome rearrangements, potentially triggering neurodegeneration and senescence [9][10][11]. XRCC1 acts as a molecular scaffold, binding repair proteins at damaged regions, while PARP1 labels damaged DNA for recruitment of DDR factors [12][13]. Reduction in XRCC1 expression might inhibit highly error-prone MMEJ [10][14] and redirect the repair efforts towards less error-prone NHEJ pathways, whose genes are highly transcribed in NMRs [15]. Moreover, high PARP1 expression was previously correlated with lower recombination rates [14], which in turn might protect from chromosomal rearrangements, oncogene activation, or the development of Alzheimer’s disease [16].
The NMR has three copies of CCAAT enhancer binding protein gamma (CEBPG) [8]—a transcription factor (TF) that activates antioxidant and DNA damage repair genes. Increased copy number of CEBPG may be implicit in higher basal levels of antioxidants [17][18], which in turn safeguard cells from reactive oxygen species and correlate with greater neuroprotection [19]. RPA4 is an analogue of RPA2, which binds single-stranded DNA (ssDNA) during repair and replication, found in NMR, which forms a noncanonical RPA complex (containing RPA4) that shows an increased affinity for DNA damage factors and facilitates cell cycle stalling [20]. RPA4 may allow NMR to conduct accurate DNA repair without risking cell cycle progression and conversion of ssDNA breaks to double-stranded (ds) breaks during replication as dsDNA breaks are capable of inducing senescence [21].
SOD1 activity, which is dependent on SOD1 binding to zinc ions [22][23], worsens over time as zinc homeostasis is gradually lost in ageing humans [24]. Alpha 2 Macroglobin (A2M) binds zinc and functions as a protease inhibitor and chaperone against protein aggregation [25]. This might explain NMRs’ resistance to zinc toxicity [26] and optimal zinc homeostasis throughout their lives, which might promote efficient SOD1 activity. Optimum function imparted by better zinc homeostasis, via high A2M levels, may be protecting NMRs from neurodegeneration during hypoxic conditions, which would typically trigger or exacerbate DNA damage.
NRF2 is a TF, activating over 200 genes associated with the antioxidant and anti-inflammatory response, under constant activity in NMRs [27]. KEAP1 E3 ligase, which negatively regulates NRF2 via ubiquitination, is 3X less transcribed in NMRs than mice [27]. BRCA1, also implicit in stability and activity of NRF2, shows positive selection in the protein binding region interacting with NRF2 [28]. Positive selection in a region of BRCA1 intrinsic to NRF2 stability and reduced KEAP1-dependent NRF2 degradation suggests a selective advantage for constitutive high antioxidant gene expression in NMRs.
Epigenetic rewriting is also central to induced pluripotent stem cells (iPSC) production, the class of pluripotent cells generated from differentiated somatic cells [29]. Initial, canonical iPSC induction proved unsuccessful; NMR cells required a knockdown of retinoblastoma protein (Rb) before successful iPSC induction could occur [30]. Moreover, many pluripotency related genes in mice contain bivalent domains and their chromatin landscape is euchromatic whereas NMR lacks these bivalent domains and their chromatic landscape shows extremes of compaction and relaxation across their genome [30][31]. The gradual loss of epigenetic marks in human cells contributes to ageing phenotypes, such as poor hair growth and slowed collagen production [32].
Senescence is the process of a cell cycle arrest triggered by multiple causes, including excessive DNA damage. Senescent cells release senescence-associated secretory phenotypes (SASP) which can poison the local cellular microenvironment. SASPs can block the clearance of senescent cells leading to their accumulation, which is a hallmark of age-related diseases. Research into drugs called senolytics, which help clear senescent cells from the body, has shown great promise in reducing age-related phenotypes. Moreover, the removal of senescent cells reduces SASP accumulation, which helps to return to optimum tissue function [33].
Neurons can exhibit senescent-like phenotypes impairing local tissue [34]. NMR neurones are often exposed to hypoxia, due to their low oxygen habitat, which has been shown to be toxic to human neurons [35]. Thus, the resistance of NMR neurons to functional decline and senescence-like phenotypes upon hypoxia is essential to NMRs’ longevity. Compared to mice, NMR neurones showed 4X increased survival rates 2 weeks post mechanical damage and expressed STAT3 important for remyelination, which was absent in control mice.
Schwann cells are wrapped around axons to generate the myelin sheath [36] and support axonal regeneration [37]. As NMR cells maintain STAT3 expression, they presumably do not lose the capacity for axon regeneration. Moreover, due to elevated A2M levels, NMR neurones maintain optimal zinc homeostasis, which is required for myelination—a crucial step in axon regeneration [25][38][39]. Finally, NMRs’ low caspase-3 signal suggests a weak apoptotic response to neuronal damage, which might suggest normal brain development and continual neurogenesis [40].
Astrocytes have a high glycolytic rate [41] and the presence of astrocytes improves neuronal recovery by inducing neurogenesis and angiogenesis, as ablation of astrocytes in mice inhibited neuronal repair [42][43]. Moreover, lactate can be produced from pyruvate—the end product of glycolysis. Lactate is a crucial energy source for neurones after being shuttled to them from astrocytes. Preferential F1P-driven glycolysis in NMR astrocytes may not only be used to support NMR brain function, but it also produces lactate that fuels oxidative phosphorylation during hypoxia.
Proteasome inhibition by exogenous chemicals or stress can lead to a build-up of biologically dysfunctional proteins and reduce the pool of available amino acids [44]. Heat Shock Proteins (HSPs) are stress-induced proteins acting as chaperones, ensuring correct protein structure or shuttling misfolded/damaged proteins for degradation [45]. NMRs also show much higher basal autophagy levels than mice [17] and require greater accumulation of misfolded proteins to trigger the Protein Unfolded Response (PUR) [46]. Therefore the novel NMR HSP40/HSP72 heterodimer may function as NMRs the primary chaperone during mild stress, while proteolysis and autophagy may replenish the pool of amino acids, which all together reduces ER stress and inhibits PUR response [47].
Crucially, high translational accuracy might extend cell viability. In most human cells, HIF1α is expressed constitutively at low levels, though usually degraded [48]. Due to their species-specific changes affecting HIF1α degradation, NMRs are likely to retain an active HIF1α pool to abrogate the effects of mild hypoxia. As a result, NMRs presumably require minimal time to acclimatise to fluctuating oxygen levels, reducing the window of cellular stress and avoiding stress-induced senescence.
As NMRs age, their ubiquitin-independent degradation, facilitated by 20S proteasome and ubiquitin-dependent degradation, facilitated by 26S proteasome, show a 1.5X increase in activity compared to young NMRs [49][50][51]. Moreover, age-related rise in expression of heat shock proteins (HSP) and ubiquitin ligases (UBE1, UBE2v2) was observed in NMRs [52]. Higher basal levels of heat shock proteins as NMRs age might maintain high protein flux through the proteasome, preventing build-up of dysfunctional proteins Therefore, proteasomal chaperone upregulation as NMRs age might increase flux through the proteasome (1.5X) to combat the age-associated build-up of dysfunctional proteins and key DDR proteins might abrogate the build-up of mutations.
The amino acid cysteine contains a reactive thiol group and is usually clustered around enzyme active sites, leading to inactivation upon oxidation [53]. However, NMRs show high amounts of cysteine in non-functional protein regions too [17][54]. The NMR shows far higher retention of protein activity during high ROS conditions, potentially through accessory cysteine oxidation, and no age-related increase in their overall cysteine oxidation [54], which indicates efficient repair of those oxidised protein moieties, or removal of the excessively damaged proteins [55]. Accumulation of proteomic damage could trigger cellular senescence through ER stress, potentially explaining NMRs’ low prevalence of senescent cells.
NMRs exhibit the unique phenotype of senescent cell death (SCD) [56], causing senesced cells to spontaneously die, unlike other species where they must be removed directly by the immune system [57]. Immune-independent apoptosis of senescent cells in NMRs prevents build-up of toxic SASP. The decrease in cellular recycling of biomolecules and elevated ROS upon hypoxia in NMR senescent cells may lead to the SCD phenotype. Thus, NMRs’ hypoxic environment may be working in their favour to maintain biological health through aiding senescent cell clearance.
NMR senescent cells transcriptome showed an expected increase in transcription of senescence-associated secretory phenotype (SASP) genes, but downregulation of genes in key pathways relating to SASP production and excretion [56]. NMR translation and ribosomal protein genes all demonstrated reduced transcription rates. Thus, when stress or DNA damage is sufficiently high, NMR cells are still able to enter senescence [58]. However, reduction in protein production and secretion results in lower production of SASP, protecting NMRs from their effects; NMR SCD phenotype enables effective clearance of senescent cells, abrogating their impact on the cellular microenvironment [56].
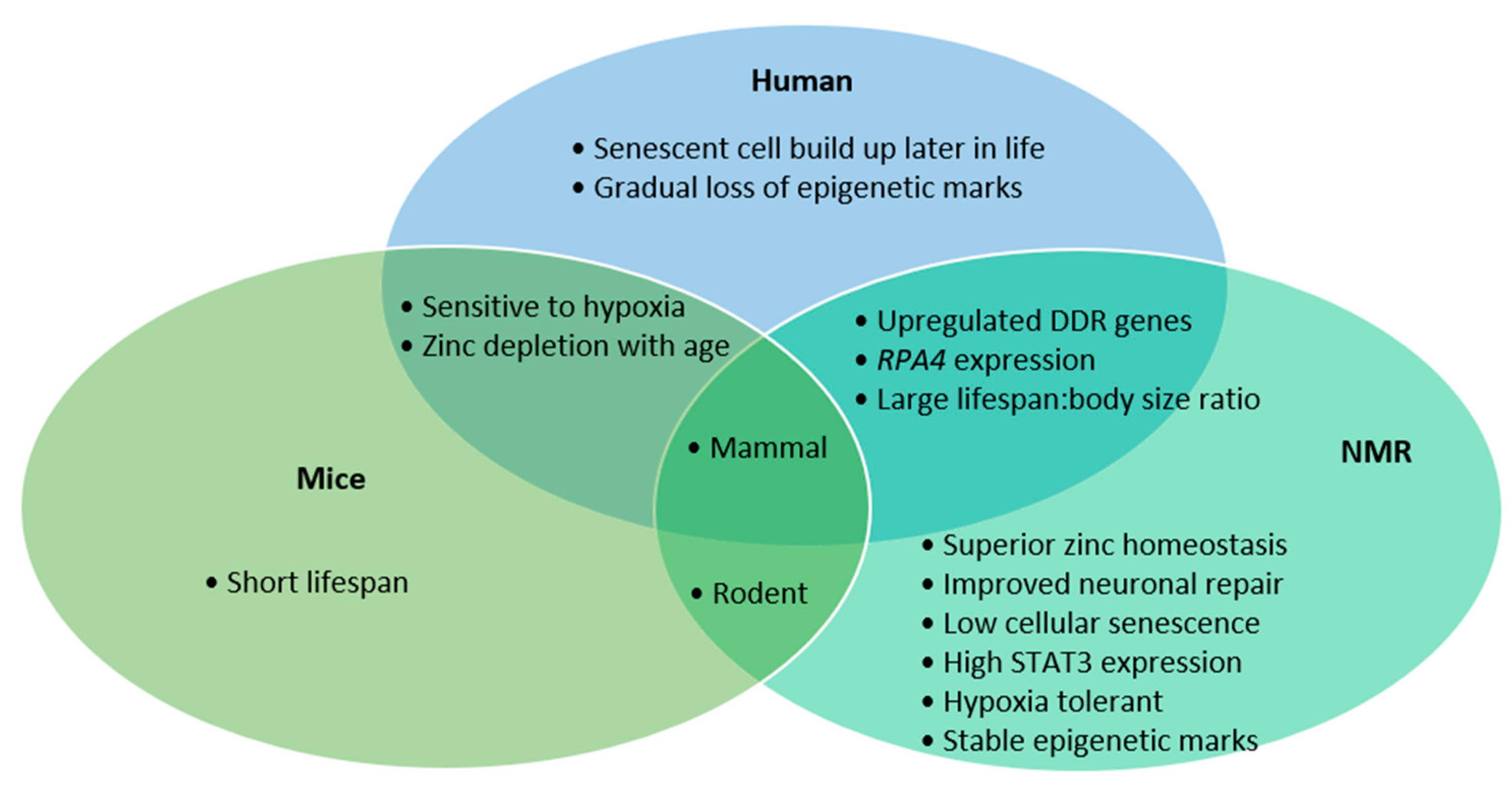
The similarities between humans and NMRs in DDR gene transcription, RPA4 presence and their lifespan compared to body-size ratio makes them perhaps a more representative model than mice (Figure 1). However, the long-life span that makes NMRs so interesting also creates a hurdle to research, due to the studies’ longitudinal nature. The NMR’s exceptional neuronal preservation is the culmination of various mechanisms, namely improved antioxidant response, higher fidelity translation, stringent DNA repair, faithful proteome function, high functioning proteasome and low prevalence of senescent cells. Though few organisms can continue indefinitely, which processes show an increased burden as NMRs age may identify novel biological targets to mitigate our own degeneration.