A central characteristic of many types of cancer is altered energy metabolism processes such as enhanced glucose uptake and glycolysis and decreased oxidative metabolism. The regulation of energy metabolism is an elaborate process involving regulatory proteins such as HIF (pro-metastatic protein), which reduces oxidative metabolism, and some other proteins such as tumour suppressors that promote oxidative phosphorylation. In recent years, it has been demonstrated that signal transducer and activator of transcription (STAT) proteins play a pivotal role in metabolism regulation. STAT3 and STAT5 are essential regulators of cytokine- or growth factor-induced cell survival and proliferation, as well as the crosstalk between STAT signalling and oxidative metabolism. Several reports suggest that the constitutive activation of STAT proteins promotes glycolysis through the transcriptional activation of hypoxia-inducible factors and therefore, the alteration of mitochondrial activity. It seems that STAT proteins function as an integrative centre for different growth and survival signals for energy and respiratory metabolism.
1. Introduction
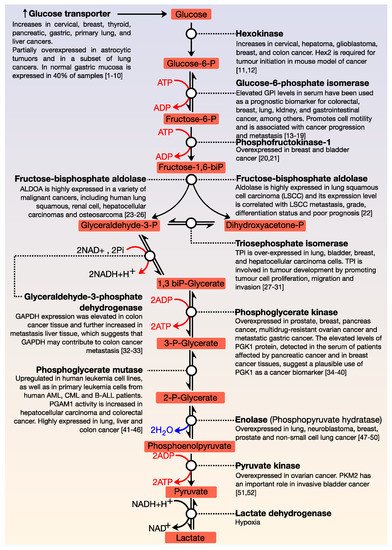
All cells need energy and for this purpose, they use macromolecules, which are degraded and thus, cells obtain the necessary energy for their essential functions. The central axis of energy metabolism consists of glycolysis, the Krebs cycle, and the respiratory chain. Glucose is the primary fuel and oxidises, via glycolysis, to provide energy for all cells (). This metabolic pathway consists of 10 consecutive enzymatic reactions that convert glucose into two molecules of pyruvate, which connect with other metabolic pathways. During glycolysis, two net ATP and two net NADH molecules are produced. This pathway usually works with limited amounts of oxygen and is only an effective means of energy production during short, intense exercise, providing energy for a period of 10 s to 2 min (it is dominant for about 10–30 s during maximal effort). A by-product of glycolysis is lactic acid; this molecule accumulates in muscles and can produce tiredness and soreness. ATP is the primary energy source for performing metabolic work, while NADH can have different functions—it is the source of reducing power in anabolic reactions or, in the presence of oxygen, it can be oxidised in the respiratory chain.
Figure 1. General view of glycolysis. The main steps of the regulation of this pathway are the conversion of glucose to glucose-6-phosphate; fructose-6-phosphate to fructose 1, 6-biphosphate; and the formation of pyruvate from phosphoenolpyruvate. All glycolytic enzymes are deregulated in cancer.
The functions of glycolysis are in the generation of high energy molecules (ATP and NADH) as a source of cellular energy for aerobic respiration (in the presence of oxygen) and fermentation (without oxygen); the pyruvate generated in glycolysis enters the Krebs cycle and the intermediates of three and six carbons can be used in other metabolic processes. As shown in , each reaction in glycolysis is catalysed by a specific enzyme. Many enzymes of the glycolytic pathway play significant roles in diverse non-glycolytic processes, such as transcriptional regulation, which enables cancer cells to meet other cellular demands. All enzymes that are involved in this pathway are deregulated in cancer
[1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52].
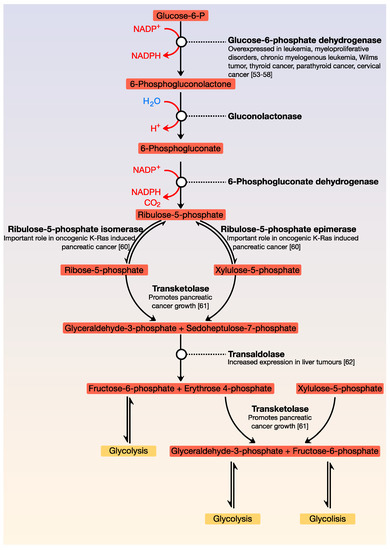
A critical metabolic pathway that is parallel to glycolysis is the pentose phosphate pathway (also called the phosphogluconate pathway or hexose monophosphate shunt). It generates NADPH as well as ribose-5-phosphate; this molecule is a precursor for the synthesis of nucleotides. There are two distinct phases: the first is oxidative, in which NADPH is generated, and the second is the non-oxidative synthesis of 5-carbon sugars. This pathway is critical for cancer cells because it generates pentose phosphates, which support a high rate of nucleic acid synthesis. It also provides NADPH for both the synthesis of fatty acids and cell survival under stressful conditions with high levels of intracellular reactive oxygen species. There is increasing evidence that cancer cells modulate the flux of the hexose monophosphate shunt for their benefit since alteration of the pathway contributes directly to cell proliferation and survival. Like glycolysis, most enzymes of the pentose pathway are deregulated in cancer ()
[53][54][55][56][57][58][59][60][61][62].
Figure 2. General view of the pentose phosphate pathway. This pathway is parallel to glycolysis and is the main source of NADPH and pentoses. Its point of regulation is the conversion of glucose-6-phosphate to 6-phosphogluconolactone.
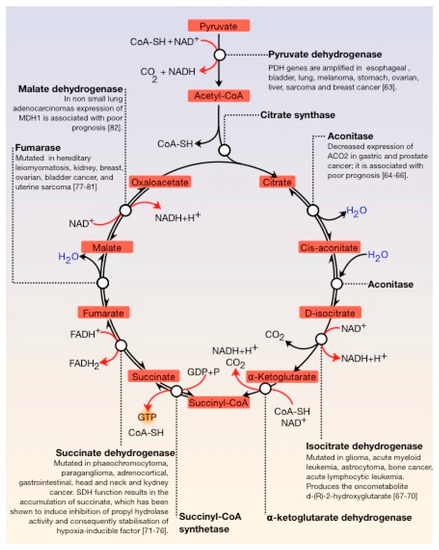
The Krebs cycle, also known as the tricarboxylic acid cycle (TCA) or citric acid cycle (CAC), is fundamental in all cells that use oxygen during cellular respiration. Furthermore, it provides precursors (for example, ketoglutarate and oxaloacetate) for the production of amino acids as well as other fundamental molecules. The TCA cycle is composed of a series of enzymatic reactions occurring in the mitochondrial matrix and is a vital source of metabolic intermediates, providing energy, macromolecules, and redox balance to the cell. This is important for highly proliferating cells, like tumour cells, which require a continuous supply of small metabolites for the synthesis of proteins, nucleic acids, and lipids. Aberrant TCA cycle function is implicated in a wide variety of pathological processes such as obesity. Moreover, several TCA cycle enzymes are deregulated in cancer ()
[63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82].
Figure 3. General view of the Krebs cycle. The regulation points are the condensation of oxaloacetate with acetyl-CoA, the conversion of D-isocitrate to α-ketoglutarate, and the conversion of this molecule to succinyl-CoA. Several enzymes of the tricarboxylic acid (TCA) cycle are deregulated in cancer. Several enzymes of the tricarboxylic acid (TCA) cycle are deregulated in cancer.
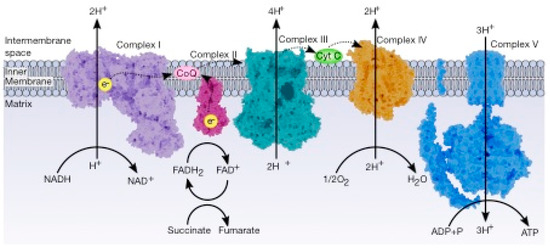
The electron transport chain (ETC) controls combustion, which generates energy to be used to drive the oxidation of NADH, which produces three ATP molecules and FADH2 (which produces two ATP molecules) at the expense of reducing oxygen. The ETC is a series of sequential redox reactions in which the electrons are transported from one component to the next to reach the final acceptor, where the electrons reduce the oxygen to water. The phosphorylation of ADP to produce ATP is carried out by ATP synthase; this process is called oxidative phosphorylation and is the way that aerobic organisms obtain energy ().
Figure 4. General view of the electron transport chain. This pathway is a series of five complexes transferring electrons from donors (NADH or FADH2) to acceptors (final acceptor is O2) and generates an electrochemical gradient which drives the synthesis of ATP.
2. Role of the Transcription Factors STAT and HIF in the Deregulation of Energy Metabolism in Cancer Cells
2.1. Role of STAT Proteins in Metabolism
Signal transducer and activator of transcription (STAT) proteins are essential transcription factors for the cellular response to cytokines and growth factors
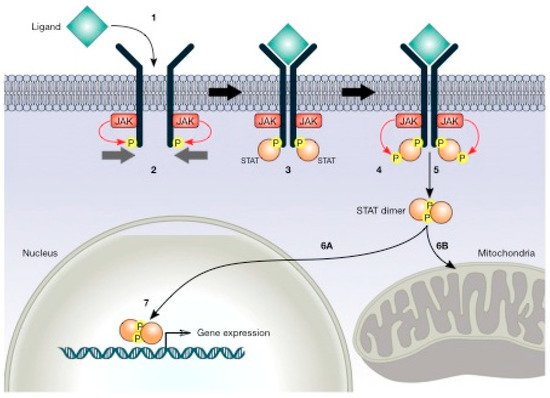
[83][84][85]. Upon the binding of a ligand, the receptor becomes phosphorylated in tyrosine (pTyr); STAT proteins bind to the receptor and are phosphorylated by JAK kinases. They then dimerise, translocate into the nucleus, and regulate the expression of genes that modulate cellular functions (). Increasing evidence suggests that STAT signalling may be involved in the regulation of cellular metabolism
[86].
Figure 5. JAK/STAT (signal transducer and activator of transcription) pathway. This pathway initiates with ligand binding (1), which induces receptor dimerisation, and the associated JAKs phosphorylate the receptors (2). STATs bind to the phosphorylated receptor (3). Subsequently, JAKs phosphorylate STATs (4). STATs separate from the receptor and dimerise (5) and then migrate to the nucleus (6A) or mitochondria (6B). STATs regulate gene expression in the nucleus (7).
STAT molecules (particularly STAT3 and STAT5) are constitutively activated in a large variety of cancers. The actual model describes STAT molecules regulating the expression of nuclear target genes involved in proliferation, cell cycle progression, and resistance to apoptosis
[86]. In Ras-mediated oncogenesis, STAT3 has been reported in mitochondria, and altered glycolytic and oxidative phosphorylation activities have been suggested, indicating that STAT proteins regulate an unknown metabolic function in mitochondria
[87]. These results suggest that STATs might have supplementary functions in the mitochondria to alter cellular metabolism in ways that favour oncogenic transformation
[86]. We discuss how the transcription factors STAT3 and STAT5 participate in the regulation of energy metabolism and their involvement in the regulation of HIF-1α, an important regulator in the cancer hypoxic microenvironment.
2.2. STAT3
The STAT3 transcription factor is well known for functioning as an anti-apoptotic factor, enhancing DNA repair in several malignancies, like leukemic stem cells
[88], breast
[89][90], prostate
[88][89], fibrosarcoma, myelomas, lymphomas, head and neck, lung, pancreatic
[90][91], and glioblastoma
[92]. STAT3-dependent gene expression in cancer is heterogeneous, reflecting the implication of this factor in multiple steps of the oncogenic program
[93]. In response to interferon β, pTyr-STAT3 participates in modulating mitochondrial electron transport chain (ETC) activity and oxidative phosphorylation
[93]. Some observations suggest the existence of an essential correlation between STAT3 and cell metabolism; the latter process is aberrantly regulated in cancer cells. Evidence exists that constitutive activation of STAT3 promotes glycolysis through transcriptional induction of HIF-1α and decreased mitochondrial activity
[94][84]. Activation of STAT3 precedes HIF-1 transcriptional response in oxygen deprivation; pSTAT3 has a peak after five minutes of oxygen deprivation, since maximum HIF-1α stabilisation requires 120 min
[95]. In oesophageal squamous cell cancer, STAT3 bound to the HIF1α promoter and the knockdown of STAT3 inhibited epithelial–mesenchymal transition and downregulated HIF1α
[96]. In malignant peripheral nerve tumours, STAT3 and HIF-1α promote oncogenic phenotypes, similarly to oesophageal cancer, where STAT3 knockdown was sufficient to block the expression of HIF-1α
[97].
Exactly how STAT3 phosphorylation can be regulated within the mitochondria is not understood, however the phosphatase SHP2 was proposed as a potential player in dephosphorylating mitochondrial STAT3
[98]. Phosphorylation in serine 727 (S727) of STAT3 has emerged as a critical regulator of metabolic processes. STAT3 is localised in the mitochondria and some reports indicate that the molecule GRIM19, a component of Complex I of the ETC that was previously identified as a protein capable of interacting with STAT3, is necessary for the import of STAT3 into the mitochondria
[99]. Inside the mitochondria, STAT3—phosphorylatedat S727—can interact with the Complexes I and II, which results in a reduction of reactive oxygen species (ROS)
[86][100]. This function is essential under some stressful conditions, such as cardiac ischemia. In this case, the mitochondrial localisation of STAT3 protects the cells by preserving the activity of Complex I, reducing ROS production and the activation of caspase 3
[101]. STAT3 also interacts with cyclophilin D in the mitochondrial matrix, preventing the opening of the mitochondrial permeability transition pore (MPTP), avoiding apoptosis induced by calcium and necrosis
[93]. STAT3 is capable of maintaining Ras-mediated carcinogenesis due to increased aerobic glycolysis and ETC activity, diminished opening of MPTP, and the lack of STAT3, which decreases the activity of Complex V (ATP synthase) dramatically
[100].
Evidence exists that STAT3 affects the optimal activity of the ETC because it can modulate the energy status of the cell. For example, in pro-B cells and in murine hearts, mitochondria increase the activity of Complexes I and II
[100]; in MEF cells, expressing the mutant H-RasV
12, STAT3 increases the activity of Complexes II and V
[87]; in MEF cells lacking SIRT1 (
SIRT1 KO), they show an increase in the activity of Complexes I, III, and IV and a decrease in Complex II activity
[102]; in the murine cardiomyocyte cell line HL-1, this transcription factor elevates the activity of Complexes I–IV
[103]. It seems that STAT3 functions as an integration centre for different survival and growth signals through its nuclear and mitochondrial activity
[93]. Furthermore, the relative concentration of this molecule in the organelle is low; biochemical fractions reveal that 5–10% of total STAT3 localises in the mitochondria of multiple tissues and cell lines
[100]. Nevertheless, how STAT3 carries out its activities within the mitochondria remains an open question.
2.3. STAT5
STAT-5 is considered to be an oncogene because it brings about the activation of cyclin D1, c-Myc, and Bcl-xl expression and is involved in promoting cell cycle progression, cellular transformation, and in preventing apoptosis
[104]. Aberrant signalling of STAT3 and STAT5 is present in different solid tumours, like bladder, breast, colon, head and neck, cervical, and liver cancers, gliomas, melanomas, and haemopoietic malignancies
[105][106][107][108]. Cholez et al. gave one of the first pieces of evidence linking STAT5 to oxidative metabolism (in pre-B cells). They used a proteomic approach to identify differential types of regulation in cells either expressing or not expressing a dominant negative form of STAT5A (NALM6Δ5). Among the 14 identified proteins, six were involved in the control of oxidative stress. The possible link between STAT5 and oxidative metabolism could be in the downregulation of transaldolase, glutathione synthetase, DJ-1, and thioredoxin domain-containing protein 9 (proteins 1–4) expression
[109]. The downregulation of the levels of DJ-1 protein (known as Parkinson disease protein 7) in pre-B cells may, therefore, increase cell death susceptibility through oxidative metabolism and the upregulation of QPRTase (quinolatephosphoribosyltransferase) and DDAH2 (dimethylargininedimethylaminohydrolase). Some of these proteins control the levels of ROS
[109]. Cancer cells could use this mechanism to modify their metabolism and adapt better to the tumour microenvironment.
The JAK/STAT pathway (in particular, STAT5) could regulate glucose metabolism by driving the expression of Pyruvate dehydrogenase kinase (PDK). STAT5 mediates the expression of PDK4 in adipocytes in response to prolactin
[110]. Chueh et al. demonstrated an interaction between STAT5 and a metabolic enzyme, PDC-E2, in the mitochondria. STAT5 exhibits unique DNA-binding activity. The presence of mitochondrial STAT5 in tumour cells and cytokine-stimulated cells also coincides with their metabolic shift towards aerobic glycolysis
[86]. In a later report, the same group demonstrated that PDC-E2 might function as a coactivator in STAT5-dependent nuclear and mitochondrial gene expression. The model proposed by Chueh et al. for nuclear–mitochondrial crosstalk through cytokine-induced STAT5 and PDC-E2 interaction initiated by cytokine stimulation is that receptor dimerisation induces tyrosine phosphorylation of the associated JAK2 tyrosine kinase. Active JAK2 phosphorylates the cytoplasmic tails of receptor subunits. Cytoplasmic STAT5 is recruited to the receptor complex and phosphorylated in tyrosine by the JAK2 kinase. STAT5 proteins dimerise and translocate into the nucleus and the mitochondria. In the nucleus, tyrosine phosphorylated STAT5 binds to the promoter regions of distinct target genes. The associated PDC-E2 may work and collaborate with histone acetyltransferase (HAT) to enhance STAT5-dependent nuclear gene expression. On the other hand, the binding of tyrosine-phosphorylated STAT5 to the control region of mitochondrial DNA may modulate transcription initiated from the heavy strand promoter (HSP) and light strand promoter (LSP)
[110][111].
The translocation of STAT5 into the mitochondria requires phosphorylation in specific tyrosine residues. The mitochondrial localisation of tyrosine-phosphorylated STAT5 was observed in leukemic T cells (these cells express a constitutively activated STAT5), and the presence of STAT5 increased in response to IL-2. By contrast, the cells that were not treated with IL-2 did not show the presence of STAT5 in the mitochondria. Once in the mitochondria, STAT5 interacts with the E2 protein, a component of the pyruvate dehydrogenase complex; it is also able to bind to the mitochondrial genome, precisely into the D-loop region
[86]. The mitochondrial localisation of STAT5 suggests that it may be involved in mitochondrial gene regulation, also coinciding with the metabolic shift to aerobic glycolysis observed in T cells and leukaemias stimulated with cytokines. HIF-2α (a closely related HIF-1α isoform) is also a target gene of STAT5 in haemopoietic stem cells (HSC)
[112]. The decrease in the expression of HIF-2α reduces the expansion of HSC induced by STAT5. Glucose uptake is enhanced in HSC cells expressing STAT5, and in these cells, HIF-2α is required for the upregulation of genes associated with glucose metabolism. In T cells, it has also been observed that STAT5 mediates glucose uptake
[113]. Both isoforms of HIF (HIF-1α and HIF-2α) regulate the expression of numerous common genes, while HIF-1α preferentially induces genes of the glycolytic pathway
[114][115]. HIF-2α is involved in the regulation of essential genes for tumour growth, cell cycle progression, and maintenance of the pluripotency of stem cells, like the proto-oncogene c-Myc41 and the stem cell factor OCT-3/4.
Chronic liver diseases and the development of hepatocellular carcinoma are tightly related and represent a real medical challenge as treatment options are minimal. Some studies using animals have shown that the genetic deletion of STAT5 in the liver is associated with a high susceptibility to fatty liver disease, fibrosis, and cancer, pointing to a protective role of hepatic STAT5 in mouse models of chronic liver disease
[116]. Several studies have suggested that growth hormone (GH)-STAT5 signalling plays a vital role in controlling hepatic lipid metabolism. Mice with a liver-specific STAT5 ablation were shown to develop steatosis, glucose intolerance, insulin resistance, late-onset obesity, and impaired liver regeneration. Notably, the expression of genes associated with adipogenesis (PPARγ) and fatty acid uptake (CD36, or fatty acid translocase) were upregulated in Stat5a/b-deficient mice. These changes may partially explain steatosis induced by loss of STAT5
[117][118]. Barclay et al. demonstrated that deficient GH-dependent STAT5 signalling correlates with steatosis through microarray analysis, quantitative PCR, and chromatin immunoprecipitation identified putative targets of STAT5 (FA (fatty acid) synthetase, CD36 signalling) that are responsible for the steatosis associated with a healthy diet
[117]. The liver is a target organ for steroid hormone metabolism. Several studies have suggested that GH-STAT5 regulates genes linked to steroid metabolism. One of these genes,
HSD3b5, catalyses the formation of the poorly active metabolite androstanediol from dihydrotestosterone
[118].
HSD3b5 gene expression was shown to be downregulated in the livers of STAT5-deleted male mice
[119][120]. By contrast, an alternative gene involved in testosterone metabolism, testosterone 16α-hydroxylase (Cyp2b9), which hydroxylates testosterone at the 16α position, was found to be upregulated in STAT5-deleted male mice. Cyp7b1 (oxysterol 7α-hydroxylase), which is responsible for the hydroxylation of dehydroepiandrosterone, androstenediol, and 25-hydroxycholesterol, was found to be downregulated in STAT5-deleted male mice
[121].
Another critical organ in which the STATs play an essential role is the heart; JAK1, JAK2, TYK2, and all the members of the STAT family are expressed in the heart
[122]. Multiple studies have demonstrated a favourable and protective role of STAT3 in the heart. This role has mainly been pointed out using data from animal experiments. As STAT3 knockout mice were shown to have early embryonic lethality
[123], specific cardiac myocyte
STAT3 knockout mice have been a helpful tool to investigate the role of STAT3 in the heart. The use of these mice and the pharmacological inhibitor of JAK2 (AG490) demonstrated a protective and anti-apoptotic role of STAT3. This role has mainly been demonstrated using a model of ischemia/reperfusion injury
[124]. Similar cardio-protective effects have been described for STAT5 activation
[125]. Heusch et al. used a model of cardio-protection by remote ischemic preconditioning (RIPC), and only STAT5 activation was associated with protection by RIPC. The available data suggest that the functions of STAT3 and STAT5 might have different roles in pigs and humans; STAT5, but not STAT3 activation, is associated with protection in human hearts, whereas STAT3 activation and possibly STAT5 inhibition are associated with protection in pigs. Alternatively, different procedures and protocols of cardio-protection may recruit a different pattern of STAT isoform signalling
[125]. Tumour cells could use this mechanism to evade apoptosis and increase their resistance to stress caused by a low oxygen concentration. STAT5 undergoes nuclear and mitochondrial activities, and both functions cooperate to induce metabolic shift, however the role of this transcription factor in the mitochondria is unknown and remains an open question.
As mentioned above, the STATs are essential for promoting glycolysis through the activation of HIF molecules. For example, STAT3 induces the expression of HIF-1α, augments glycolysis, and decreases mitochondrial activity. Furthermore, STAT5 can induce the expression of HIF-2α, which is necessary for the upregulation of genes associated with glucose metabolism. Thus, it is clear that STAT proteins are involved in regulating hypoxia-inducible factors that are the main regulators of oxygen homeostasis. Hypoxia is a critical microenvironmental factor that is related to progression and metastasis. Most of the solid tumours show hypoxic conditions, and this minor concentration of O
2 induces an altered regulation in gene transcription, leading to changes in cell metabolism
[126][127].
2.4. The Importance of Oxygen and HIF-1α in the Regulation of Metabolism
Oxygen is a basic necessity for life as it is the final acceptor of electrons in the ETC; its relative abundance or absence can modify processes like embryogenesis, wound healing, and stem cell maintenance
[128][129][130][131]. In fact, because of its physiological importance, oxygen levels and molecular pathways regulated by oxygen are strictly regulated, so failure in the proper regulation in response to low oxygen (hypoxia—an oxygen concentration of approximately 6% or lower generates hypoxic stress) can promote a number of diseases, including diabetic retinopathy, ischemic heart disease, and cancer
[128][132][133]. In cancer, the microenvironment of tumours is related to chronic hypoxia
[133][134]. In response to low levels of O
2, the tumour cells turn on specific genes that promote adaptation to hypoxic stress, including those involved in angiogenesis, cell survival, proliferation, evasion of growth suppressors, metastasis, metabolic reprogramming, and mortality
[135][136]. It is well documented that hypoxia promotes tumorigenesis and contributes to a poor clinical prognosis
[137][138].
Hypoxia inducible factor-1alpha (HIF-1
α) is a critical molecule that is overexpressed in many types of cancer; this transcription factor forms heterodimers consisting of an oxygen-dependent subunit (subunit α) and an independent oxygen subunit (subunit β). There are three isoforms of the α subunit (HIF-1α, HIF-2α, and HIF-3α) and three isoforms of the β subunit, also known as the aryl-hydrocarbon receptor nuclear translocators (Arnt1, Arnt2, and Arnt3). Subunit α translocates into the nucleus and under normal conditions, has an extremely short half-life—less than five minutes
[139]. The degradation of this subunit is mediated by the oxygen-dependent degradation domain (ODDD) positioned within the N-TAD, which contains specific proline residues (Pro402 and Pro564 in HIF-1α and Pro405 and Pro531 in HIF-2α) that are hydroxylated in an average oxygen concentration by a particular group of prolyl hydroxylases (PHDs)
[140]. However, the α-subunit can dimerise with the β-subunit (this subunit is insensitive to oxygen and hence, is constitutively expressed) when the oxygen concentration is under 6%
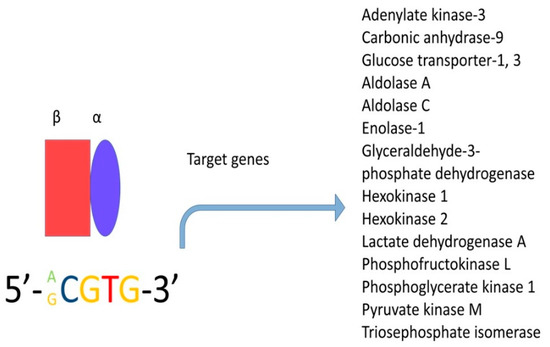
[140][141]; this complex binds to consensus sequence 5′-RCGTG-3′, which is present in the enhancer of the hypoxia response element ().
Figure 6. Hypoxia-inducible factor (HIF) is a transcription factor and is expressed in all metazoan organisms. This factor is composed of two subunits: HIF-1α and HIF-1β. Under hypoxic conditions, HIF regulates the expression of hundreds of genes. In this figure, only the genes related to metabolism are shown.
High levels of expression of HIF-1α are associated with high mortality in cancer
[142]; HIF is capable of inducing transcription of the pyruvate dehydrogenase kinases (PDK) 1 and 3
[143][144]. The PDK phosphorylates and inactivates the pyruvate dehydrogenase (PDH), preventing pyruvate from entering into the Krebs cycle, hence reducing mitochondrial oxygen consumption and decreasing the production of reactive oxygen species (ROS). HIF promotes the conversion of pyruvate to lactate through lactate dehydrogenase. This enzyme is a tetramer that includes the H subunit (LDH-H; this subunit is expressed ubiquitously by the gene IDHB). The LDH-H that is found in the heart is more active at low concentrations of pyruvate and is strongly inhibited by an excess of pyruvate (approximately 10
−2 M). The subunit M (LDH-M, expressed by the gen IDHA) maintains its activity at relatively high pyruvate concentrations
[145]. The IDHA gene is a direct target of HIF and is highly inducible by hypoxia. HIF, therefore, promotes the formation of a complex formed exclusively by LDH-M, which is more efficient in converting pyruvate to lactate; this results in a decreased flow of pyruvate into mitochondria
[143].
HIF-1α is expressed in cervical cancer, which represents an early event in tumour development and is expressed in both normoxia and hypoxia conditions. The levels of glucose transport protein 1 (GLUT1) gradually increases during the transition from normal cervix conditions to cervical intraepithelial neoplasia; the expression of this molecule is related to lymph node metastasis, and the expression levels of GLUT1 and HIF-1α are correlated, indicating that HIF-1α may regulate the expression of downstream genes that are involved in the energy supply
[146]. On the other hand, the contribution of HIF-1α to the regulation of hypoxic cell survival or death remains controversial. It has been reported that this molecule can have either pro-apoptotic or anti-apoptotic effects in different systems
[147]. During severe hypoxia (or anoxia), apoptotic cell death is crucial to avoid hypoxia-induced mutations in cells. One proof of hypoxia-induced apoptosis is the suppression of the electron transport chain on the inner membrane of mitochondria
[148]. Reports exist that HIF-1α can initiate hypoxia-mediated apoptosis by enhancing the expression of several genes (Bcl-2, p53, and others)
[149]. However, during hypoxia, the translocation of the Bax protein to the mitochondria is downregulated and the inhibitor of apoptosis protein 2 (IAP-2) levels are upregulated; this combination of effects preserves the mitochondrial integrity and may help with cell survival during hypoxia. HIF-2 also plays an essential role during hypoxia by positively regulating cell proliferation, which is controlled by Myc
[150][148]. In fact, during severe or prolonged hypoxia, the vast majority of the cells undergo apoptosis; nevertheless, some of the cells adjust to the environment and survive by avoiding necrosis and apoptosis, therefore resulting in an aggressive phenotype. This phenomenon suggests that cells that are non-sensitive to apoptosis in a tumour will be resistant to anticancer treatments
[151][152].
 +1 point
+1 point