Formaldehyde (FA) is a highly reactive substance that is ubiquitous in the environment and is usually considered as a pollutant. In the human body, FA is a product of various metabolic pathways and participates in one-carbon cycle, which provides carbon for the synthesis and modification of bio-compounds, such as DNA, RNA, and amino acids. Endogenous FA plays a role in epigenetic regulation, especially in the methylation and demethylation of DNA, histones, and RNA.
1. Introduction
Due to the longevity of the human population, aging and aging-related problems have become one of the most important health problems in the world. Age-related cognitive impairment (ARCI) is a predominant syndrome of aging that has attracted increasing attention in the past few decades. Although numerous hypotheses about the mechanisms and risk factors associated with ARCI development have been suggested, only a few effective therapies are clinically available
[1]. One of the potential factors contributing to ARCI is dysmetabolism of formaldehyde (FA). FA has also been suggested to be implicated in the development of Alzheimer’s disease (AD), since elevated FA levels in AD patients and animal models of AD are associated with impaired cognitive abilities. In 1999, Luo et al. investigated the effect of aldehydes, such as acetaldehyde and glutaraldehyde on Tau phosphorylation and aggregation
[2]. In 2001, Yu hypothesised that cerebrovascular semicarbazide-sensitive amine oxidase is involved in the pathogenesis of Alzheimer’s disease (AD) and vascular dementia
[3]. Later, FA was found to cause protein Tau dysfunction and aggregation, leading to cytotoxicity
[4][5][6]. Moreover, clinical investigations have shown that dysmetabolism of endogenous FA may be a risk factor for the development of ARCI
[7]. Further investigation has contributed to elucidate the pathological relationship between the dysmetabolism of FA and ARCI, as well as the early clinical symptoms of AD
[8][9][10][11].
The release of FA by drug demethylation was first described by Axelrod in 1956
[12]. In 1989, DNA methylation was considered a molecular mechanism of cell memory
[13][14]. The one carbon cycle is an essential metabolic hub in the cell that releases FA
[15]. In 1999, Tohji et al. suggested an association between DNA methylation and neurodegeneration, postulating that the decrease in methylcytosine in the region comprised between base pairs positions −224 to −101 of the amyloid precursor protein (APP) gene promoter in human cerebral cortex autopsy samples was associated with age
[16]. Afterwards, it was discovered that the methylation status of cytosines in the
TAU gene promoter region changes with aging, resulting in a downregulation of the transcriptional activity of this gene in the human brain
[16]. Tong et al. further elucidated the effect of FA on epigenetic changes associated with ARCI, and reported a relationship between FA and memory, as well as between FA and the extent of DNA and histone methylation
[17]. Later, they identified a relationship between memory loss and FA due to an imbalance in DNA methylation and demethylation
[18]. Furthermore, they also determined that high concentrations of FA related to age interfere with the function of DNA methyltransferase, leading to memory loss in AD
[19]. In 2017, the relationship between FA and cognition was systematically investigated and elucidated, including the mechanisms of FA synthesis, metabolism, and its role in cognitive impairment, which involved epigenetic mechanisms
[20]. Epigenetic alterations and the involvement of FA in memory loss suggest that the exploration of FA exposures as a risk factor may provide an insight into the pathological mechanisms of AD
[21].
2. Formaldehyde
As the simplest carbonyl compound, FA has two hydrogen atoms, one oxygen atom, and a carbon atom in the middle (CH
2O). However, unlike other simple carbon compounds, FA is reactive and can exist in several forms, such as monomeric FA, trioxane, and paraformaldehyde
[22]. FA in the upper atmosphere contributes approximately to 90% of the total environmental FA. Atmospheric FA is produced through the action of sunlight and oxygen on atmospheric methane and other hydrocarbons, after which it becomes part of the smog. In forest fires, car exhaust, and tobacco smoke, FA is an intermediate product in the oxidation of methane, and can be oxidized to formic acid
[23]. Although FA is colorless, pungent, and toxic, it is an important precursor of many materials and chemical compounds, mainly used in particle boards and coatings, as well as in other industries. FA poses a significant danger to human health, since it presents toxic
[24] and carcinogenic characteristics
[25]. Ingestion or inhalation of FA has been reported to be harmful to humans, causing eye irritation, headache, difficult breathing, leukemia, cancer, or even death
[26][27]. Thus, the use of FA is highly regulated in numerous countries worldwide.
In mammalian organisms, FA is ubiquitous and is an essential intermediate in cellular metabolism. Humans quickly convert FA into formic acid, inhibiting the accumulation of FA in the body
[28]. However, humans metabolize FA slower than rodents. The concentration of FA in the blood of human is ~0.1 mM
[29]. Endogenous FA refers to that produced in various metabolic pathways involving many compounds, such as sugars, lipids, proteins, and nucleic acids
[30][31][32]. For example, FA is formed during the metabolism of methanol or other methylated compounds, such as amino acids
[33]. The main enzymes that metabolize endogenous FA are mitochondrial aldehyde dehydrogenase 2 (ALDH2) and glutathione (GSH)-dependent alcohol dehydrogenase 3 (ADH3)
[34][35][36][37]. In addition, the intestinal microbiota also consume the excess of FA, combining it with tetrahydrofolic acid (THF) through the one-carbon cycle
[20]. Furthermore, biomacromolecules, including proteins, DNA, and RNA, react with the accumulated FA
[38]. Methylation/demethylation of DNA, RNA, and histones is considered one of the main ways to form and consume FA in vivo, as well as one of the main mechanisms of epigenetic regulation
[39][40][41].
3. Formaldehyde-Involved Epigenetics in Age-Related Cognitive Impairment
ARCI is characterized by problems with memory, language, thinking or judgement, and it is a typical feature of AD. Most cases of AD are sporadic, and numerous factors have been identified as risk factors, including aging, family history, diabetes, obesity, hypertension, and diet
[42]. Although the pathological mechanism of AD remains to be elucidated, two main hypotheses remain: ‘accumulation of amyloid-β’ and ‘tau pathology’. Besides these, numerous other pathways are known to be involved in the aetiology of AD, such as oxidative stress, inflammation, calcium channel disruption, and autophagy
[43]. In familial AD, mutations in
APP,
PSEN1 and
PSEN2 are known genetic risk factors
[44].
Bohnuud et al. reported that FA can cause hydroxymethylation of the pyrimidine bases in DNA and transform them into methyl groups through oxidation, silencing gene expression
[45]. Furthermore, Lu et al. found that FA can react with DNA, resulting in methylated DNA
[46], which can silence gene expression and inhibit DNA synthesis in cells
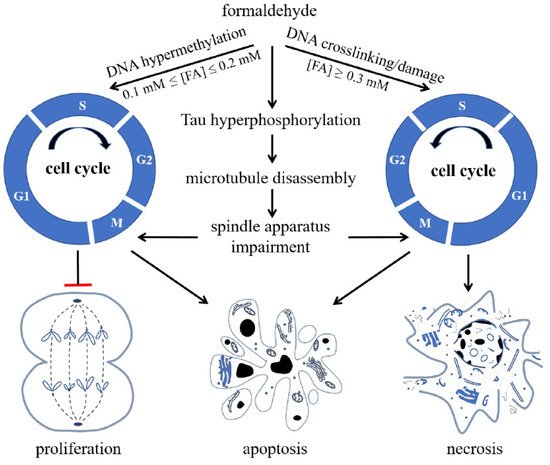
[47][48]. When the FA concentration increases, it acts on nuclear DNA, aggravates the degree of DNA methylation, and then inhibits DNA unchaining, replication, and synthesis. Hence, high concentrations of FA cause cell cycle arrest, eventually triggering apoptosis and necrosis ()
[49].
Figure 1. Formaldehyde (FA) inhibits cell proliferation and induces cell apoptosis and necrosis via its effect on cell cycle. FA induces hypermethylation of DNA when 0.1 mmol/L ≤ (FA) ≤ 0.2 mmol/L, and causes crosslinking or damage of DNA, arresting cells at S phase and restraining cells from entering G2/M phase when (FA) ≥ 0.3 mmol/L, reproduced with permission from Miao et al.
[9].
ARCI originates from chronic synaptic dysfunction
[50] and neuronal loss
[51], which are accompanied by the formation of senile plaques, which are extracellular deposits of amyloid-β (Aβ), and neurofibrillary tangles, which are paired helical filaments of hyperphosphorylated Tau. Excessive FA induces hyperphosphorylation of Tau, resulting in microtubule disassembly and damage of the spindle apparatus. In SH-SY5Y cells, these effects cause cell cycle arrest at the G2/M phase. Excessive FA also has an aberrant impact on DNA
[52]. Animal experiments have shown that methanol, which is converted to FA in vivo, can induce amyloid β deposition, Tau hyperphosphorylation, and cognitive impairment in monkeys
[53][54]. In patients with AD, the concentration of Aβ in the CSF is decreased, indicating an increase in Aβ deposition in brain tissue
[55]. In
Rhesus monkeys, FA concentration in CSF increases with aging and is negatively correlated with Aβ concentration
[56], further supporting a link between FA concentrations and Aβ accumulation.
Fei et al. reported oxidative demethylation at Ser8/26 of Aβ induces FA generation. According to their results, FA interacts with the Lys28 in the β-turn of Aβ monomers, enhancing Aβ oligomerization
[57]. Furthermore, Aβ inhibits FDH activity, leading to FA accumulation. Therefore, excessive FA and Aβ oligomers form a positive feedback cycle that may play a role in the development of ARCI. FA scavengers such as resveratrol, NaHSO
3, or coenzyme Q10 have been reported to reduce Aβ aggregation, ameliorate neurotoxicity, and improve cognitive performance in APP/PS1 mice
[10][57]. The direct interaction between FA and Tau protein methylation is yet to be investigated.
These results suggest that FA may be a risk factor for sporadic ARCI, although it is not present in all patients with AD. Endogenous FA is closely related to ARCI in patients with AD
[7]. High endogenous FA levels have been reported in patients with AD in comparison with age-matched participants with normal cognition
[58]. Moreover, exogenous FA exposure have been shown to induce AD-like changes in murine brain
[59]. Endogenous levels of FA could be employed as a diagnostic criterion for ARCI in the preclinical and clinical stages, and for epidemiological investigation of AD. The mechanisms involved in the dysmetabolism of FA in ARCI include FA-induced amyloid peptide aggregation, tau pathology, inflammation, oxidative stress, neurotoxicity, and gut microbiota imbalance. However, methylation or demethylation of DNA, RNA, histone, and amino acids are typical features of epigenetic regulation of the disease.
Recent studies have indicated that epigenetics contribute to the development of ARCI. Altered DNA methylation, histone modifications, and ncRNA interactions are characteristics commonly found in AD. According to the epigenetic clock theory of aging, epigenetic changes, including DNA methylation/demethylation, are involved in aging
[60][61][62]. In patients with AD, global DNA methylation in brain samples is decreased. Moreover, an altered DNA methylation landscape in CpG islands of susceptible genes in AD, such as
ABCA7 and
BIN1, have been shown to be associated with AD pathology
[63]. Lower levels of 5mC and 5hmC have also been reported using in vitro models of AD and in brain tissue of patients with AD
[64]. In addition, alterations in the gene expression level of HDACs have been linked to AD
[65]. Excessive histone methylation has also been reported in patients with AD
[66]. Finally, ncRNAs, including miRNAs and lncRNAs, were also found to be differentially expressed in AD pathology
[67][68].
Imbalance of Formaldehyde Metabolism Is Associated with Age-Related Cognitive Impairment
In physiological conditions, FA is metabolized and homeostasis is maintained
[49]. An imbalance in FA metabolism may be related to the development of ARCI
[20]. We believe that one of the main causes of cognitive impairment may be excessive production and accumulation of endogenous FAs in the brain. Although either a lack of or excess of FA resulting from an imbalance in metabolism should be harmful to cognitive ability, the lack or low concentration of endogenous FA is not commonly considered as a major risk factor. This viewpoint is based on the following observations: (1) Endogenous FA originates from several metabolic pathways. If less FA is produced in one metabolic pathway, the production of FA can be compensated for by other metabolic pathways
[20]. (2) The degradation pathways of FA, involving enzymes such as ADH and ALDH, are less abundant than those of the production pathways
[69]. (3) High levels of FA were observed in the digestion contents in the cecum of 7-month-old APP/PS1-transgenic mice. However, a marked increase in FA was not observed in the cecum contents of wildtype 7-month-old C57 mice
[70], nor in 24-month-old wildtype 129S2/SvPasCrl mice though their intestinal length extended with aging
[71] (
Figure S1, Permit Number: SYXK2019-06). This suggests that FA metabolism in vivo may also be related to the intestinal microbe flora. (4) Methylation and demethylation play critical roles in the catecholamine metabolic pathway. FA acts as a methyl donor in the catecholamine pathway through the SAM cycle
[72]. Accumulation of FA disturbs the metabolism of catecholamines, leading to norepinephrine depletion and cognitive decline
[73]. (5) Exogenous FA is ubiquitous in humans, despite its concentration
[74]. This may explain why no drug to increase FA concentrations has been developed for conditions in which FA concentrations are low. Therefore, we emphasize that FA accumulation resulting from the metabolism imbalance is associated with ARCI.
FA metabolism imbalance is associated with ARCI, as FA acts as a methyl group donor, participating in the methylation or demethylation of DNA, RNA, and histones and regulating the epigenetics landscape. Although the effects of FA on DNA methylation/demethylation have been investigated, the implications of FA in ARCI development need to be further investigated. FA levels influence the activity of DNMT and the degree of DNA methylation in the brain
[18]. Excessive FA has shown to decrease DNA methylation by disturbing the activities of DNMTs in vivo and in vitro. During spatial learning in Sprague–Dawley rats, there is an initial global DNA demethylation, followed by a re-methylation step associated with the elevation of hippocampal FA. Subsequently, hippocampal FA levels decrease and reach the baseline levels
[18]. Excessive FA concentrations could mimic the global DNA methylation decline associated with age-related spatial memory deficits observed in normal adult rats. Furthermore, global DNA methylation levels in the autopsied hippocampus of patients with AD are associated with a marked increase of endogenous FA levels
[19]. Thus, the dysregulation of DNMT related to defects in the global levels of DNA methylation could be one of the pathophysiological mechanisms underlying hippocampal-associated spatial memory impairment. According to this hypothesis, in the elderly, the degree of DNA methylation is reduced and endogenous FA accumulates, leading to the development of ARCI in the most severe cases. Hence, targeting FA imbalance might be an effective way to prevent ARCI. Although no approved drugs can be used for this purpose, some chemicals or compounds that have neuroprotective effects, including some Chinese medicinal herbs (e.g., resveratrol), physical methods, and lifestyle interventions are recommended as a treatment for ARCI.
We hypothesize that endogenous FA, including that derived from catecholamine, APP, Aβ peptide, and Tau protein, acts as an epigenetic factor in the formation or loss of memory associated with the methylation and demethylation of DNA, RNA, and histones. FA dysmetabolism has been suggested as a risk factor for ARCI, as this condition is associated with the disturbance of DNA methylation and demethylation. Further investigations on whether FA is associated with methylation and demethylation of RNA, histones and other biomolecules in ARCI should be performed in the future.
4. Conclusion
The review depicted the interplay between FA and epigenetics, involved in ARCI. Homeostasis of formaldehyde metabolism is maintained by several metabolic pathways, including the one-carbon cycle, which involves methylation of DNA, RNA, proteins, and amino acids. FA dysmetabolism, which is associated with the disturbance of methylation and demethylation. has been suggested as a risk factor for ARCI.
 +1 point
+1 point