The pathophysiology of PD relies on the degeneration of dopaminergic neurons located in the SNc (which project to the motor part of the striatum, caudate-putamen nucleus in humans), as well as cytoplasmic accumulation of α-synuclein-containing Lewy bodies
[43][52]. Based on the occurrence of
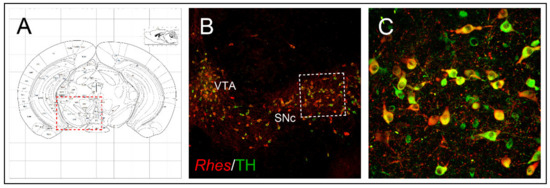
Rhes transcript in the midbrain tyrosine hydroxylase (TH)-positive neurons of SNc and VTA ()
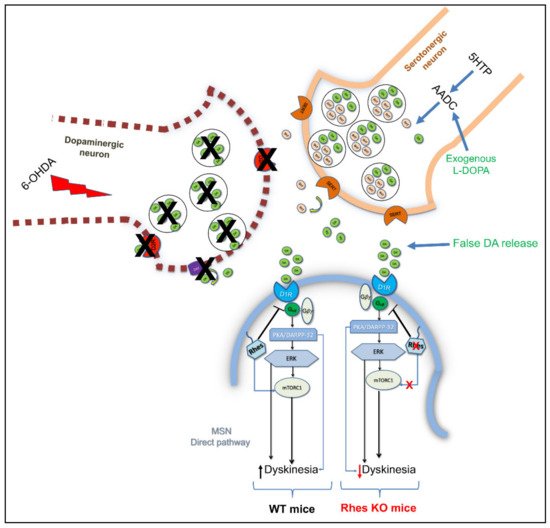
[15] and, considering its role in regulating survival-related AKT and mTOR signaling pathways, further studies sought to investigate whether Rhes could also have an impact on midbrain dopaminergic neurons’ survival, under both physiological and pathological conditions. Interestingly, lack of
Rhes led to a mild, although significant, reduction of midbrain TH-positive neurons in both 6- and 12-month-old KO male mice
[15]. As a behavioral correlate to what was observed at the morphological level, mutant male animals showed significant alterations at the beam-walking test, in an age-dependent manner, taking longer to traverse the beam, thus suggesting that Rhes might drive the nigrostriatal pathway toward a susceptibility to cell death, triggered either by aging processes or by environmental toxins
[15]. The mechanisms responsible for the PD-related neuronal degeneration are still elusive, and often controversial. However, several factors, such as gender, neuroinflammation, oxidative stress, excitotoxicity, reduced expression of trophic factors, and dysfunction of the protein degradation system, may influence the nigrostriatal pathway degeneration
[53][54]. Although several causative genes of either dominant or recessive inherited PD forms have been identified, most of them are not yet characterized, hence requiring further studies aimed at clarifying the interplay between genetics and other possible pathogenic factors
[55]. Yet, epidemiological investigations indicated that among the factors that may influence the neuronal dopaminergic degeneration, gender may have a prominent role, since males are at higher risk than females, who might be more protected by estrogens, in particular 17β-estradiol
[56][57]. Therefore, based on the involvement of the α-synuclein-mediated microglia activation in PD pathogenesis
[58][59][60], and considering the influence of Rhes upon the survival of nigrostriatal dopaminergic neurons
[15], in a recent study by Costa and colleagues, the potential role of this protein on the inflammatory response was initially investigated during the physiological brain aging, in both male and female
Rhes KO mice
[61]. Immunohistochemistry evaluations confirm the decrease in TH immunoreactivity already observed in the midbrain nigrostriatal neurons of male
Rhes KO mice
[15], but a decrease in TH immunoreactivity was also observed in female
Rhes KO mice. Interestingly, a higher number of the complement type 3 receptor (CD11b), as well as glial fibrillary acid protein (GFAP), were found in male rather than female KO mice
[61].
 +1 point
+1 point