1000/1000
Hot
Most Recent
 +1 point
+1 point
Ribosome is one of the most conserved and sophisticated macromolecular machines that carries out the essential process of protein synthesis in cells. Given the essential function of ribosome as well as difference of bacterial and eukaryotic ribosome, it has been an active target for over half clinically used antibiotics.
Ribosome is one of the most conserved and sophisticated macromolecular machines that carries out the essential process of protein synthesis in cells. All ribosomes consist of two subunits, each assembled from one or more ribosomal RNA (rRNA) molecules as well as numerous ribosomal proteins (r-proteins). The decoding center (DC) of the smaller subunit (30S in bacteria) is responsible for decoding the genetic information delivered by messenger RNA (mRNA). However, it is the rRNA of the larger subunit (50S in bacteria) that carries the main catalytic function of the ribosome—the formation of the peptide bond between the incoming amino acid attached to the transfer RNA (tRNA) in the aminoacyl- (A) site and the nascent polypeptide attached to the tRNA in the peptidyl- (P) site. In principle, the ribosome functions as an entropy trap by precisely positioning the tRNA substrates for peptide bond formation [1]. The region of the large subunit (made up of nucleosides from 23S rRNA domain V in prokaryotes) involved in the latter is called the peptidyl transferase center (PTC). Adjacent to the PTC is the opening of the tunnel through which the nascent peptide exits the ribosome.
Given the fundamental nature of protein synthesis (a.k.a. translation), it is not surprising that the ribosome is one of the main targets for chemical agents produced by certain microorganisms to give them an edge over other microorganisms in nature [2][3][4]. Furthermore, the features characteristic of bacterial translation apparatus have been historically exploited for the selection of natural antimicrobials and the development of synthetic drugs to combat bacterial infections in human and veterinary medicine as well as to benefit agriculture and food production. The discovery of antibiotics (AB) has been regarded as one of the most significant achievements in modern medicine, saving countless lives and enabling important medical procedures, including surgery and chemotherapy [5].
Unfortunately, almost as quickly as AB are adopted in clinical practice, they are in danger of becoming antiquated due to pathogens acquiring distinctive antibiotic resistance (ARE) mechanisms that have evolved in bacteria to protect themselves from “chemical weaponry” produced by themselves or rival microorganisms [2]. ARE genes can easily spread between organisms in the presence of sufficient pressure such as over- and misuse of AB in human and veterinary medicine as well as farming and food production. For instance, the AB-producing soil bacteria actinomycetes are suspected to be the origin of ARE in many other bacterial species and evidence for the exchange of ARE determinants between soil bacteria and clinical pathogens has been reported [3][6]. Therefore, it is not surprising that while there are currently numerous classes of chemically diverse AB in clinical practice that interfere with protein synthesis by binding to the ribosome (Table 1), there is an imminent threat of majority, if not all of them, being rendered obsolete due to the emergence and spread of ARE among human pathogens. Indeed, ARE mechanisms have been identified for nearly every AB currently in use in clinical practice, including virtually every ribosome-targeting AB. Table 1 summarizes the major classes of bacterial translation inhibitors termed critically important antimicrobials for human medicine (considering existing and potential ARE) by the World Health Organization (WHO) as of 2018 [7]. The WHO classification is based on AB being the sole, or one of the limited available therapies, to treat serious infections caused by pathogens that may acquire ARE genes from non-human sources in order to emphasize the importance of their appropriate use.
Table 1. Major classes of protein synthesis inhibitors grouped based on the WHO’s critically important antimicrobials for human medicine as of 2018 [7] with corresponding drug-binding sites, translation inhibition, and antibiotic resistance mechanisms.
| Antimicrobial Class | Ribosome Target and Mechanism of Action | Examples of Drugs in Clinical Use | Comments | Resistance Mechanisms |
|---|---|---|---|---|
| Highest-Priority Critically Important Antimicrobials | ||||
| Macrolides and ketolides | 50S NPET-context-dependent modulation of protein synthesis | Azithromycin § | One of few available therapies for serious Campylobacter infections and limited theraphy for MDR Salmonella and Shigella infections. Clarithromycin-resistant Helicobacter pylori causes very common infections in countries of all income levels. | Drug modification/degradation; drug efflux/membrane permeability; target mutation and modification; and target protection (ABC-F) |
| Clarithromycin § | ||||
| Erythromycin | ||||
| Josamycin | ||||
| Oleandomycin | ||||
| Solithromycin | ||||
| Spiramycin | ||||
| Telithromycin | ||||
| Troleandomycin | ||||
| High-Priority Critically Important Antimicrobials | ||||
| Aminoglycosides | 30S DC-inhibit translocation and increase error rate | Amikacin * | Sole or limited treatment of MDR tuberculosis and MDR Enterobacteriacea | Drug modification/degradation; drug efflux/membrane permeability; target mutation and modification |
| Gentamicin * | ||||
| Kanamycin | ||||
| Neomycin | ||||
| Plazomicin ¶ | ||||
| Streptomycin | ||||
| Tobramycin | ||||
| Oxazolidinones | 50S PTC (A-site)-context-dependent modulation of protein synthesis (aminoacyl-tRNA binding) | Linezolid ¶ | Limited therapy for infections due to MDR Enterococcus and MRSA | Drug efflux/membrane permeability; target mutation and modification; and target protection (ABC-F) |
| Tuberactinomycin | Subunit interface-inhibit translocation | Capreomycin | Limited theraphy for tuberculosis and other Mycobacterium infections | Drug modification/degradation; target mutation and modification |
| Highly Important Antimicrobials | ||||
| Phenicols | 50S PTC (A-site)-context-dependent modulation of protein synthesis (aminoacyl-tRNA binding) | Chloramphenicol * | One of the limited therapies for acute bacterial meningitis, typhoid and non-typhoid fever, and respiratory infections | Drug modification/degradation; drug efflux/membrane permeability; target mutation and modification; and target protection (ABC-F) |
| Thiamphenicol | ||||
| Lincosamides | 50S PTC (A-site)-inhibit peptide bond formation | Clindamycin * | ARE risk from Enterococcus and Staphylococcus aureus (including MRSA) | Drug modification/degradation; drug efflux/membrane permeability; target mutation and modification; and target protection (ABC-F) |
| Lincomycin | ||||
| Steroid antibacterials | EF-G-inhibit translation elongation and recycling | Fusidic acid | Sole or limited therapy for MRSA infections | Drug efflux/permeability; target mutation; and target protection (Fus) |
| Streptogramins A (SA) and B (SB) | SA 50S PTC (A-and P-sites)-inhibit peptide bond formation; SB 50S NPET-prevent elongation of nascent chain | Dalfopristine (SA) | ARE may result from transmission of Enterococcus and MRSA from non-human sources | Drug efflux/membrane permeability; target mutation and modification; target protection (ABC-F) |
| Quinupristine (SB) | ||||
| Tetracyclines | 30S DC (A-site)-inhibit delivery of tRNA into A-site | Doxycycline * | Limited therapy for infections due to Brucella, Chlamydia, and Rickettsia | Drug efflux/membrane permeability; drug modification/degradation; target mutation; target protection (Tet) |
| Tetracycline | ||||
| Important Antimicrobials | ||||
| Pleuromutilins | 50S PTC (A-and P-site)-inhibit peptide bond formation | Reptamulin | Only used as topical theraphy in humans | Drug efflux/membrane permeability; target mutation and modification; target protection (ABC-F) |
AB—antibiotic; ABC-F—ATB binding cassette subfamily F proteins; A-site—aminoacyl-tRNA binding site; DC—decoding center; EF-G—elongation factor G; MDR—multi-drug-resistant; MRSA—methicillin-resistant Staphylococcus aureus; NPET—nascent peptide exit tunnel; P-site—peptidyl-tRNA binding site; ¶—access group AB [8]; §—watch group AB [8]; *—reserve group AB [8].
Macrolides and ketolides are a class of ribosome-targeting drugs that bind to the 50S nascent peptide exit tunnel (NPET) adjacent to PTC and cause ribosome stalling when specific amino acid motifs are encountered at PTC and nascent chain progression is hindered [9][10]. Thus, macrolides and ketolides should be considered context-specific (depending on the nature of the nascent chain and the structure of the drug) modulators of protein synthesis. Macrolides and ketolides (foremost azithromycin, erythromycin, and telithromycin) are classified as the highest-priority clinically important microbials [7] (Table 1). Azithromycin (first or second choice treatment for chlamydia, cholera, gonorrhea, and bacterial diarrhea/dysentery) and clarithromycin (first or second choice treatment for severe pneumonia and pharyngitis) are included in the “watch group” in the WHO’s 2019 Model List of Essential Medicines naming the safest and most effective medicines needed in the healthcare system [8]. “Watch group” includes AB with a relatively high risk for the selection of ARE, whose usage should therefore be monitored and restricted to essential first or second choice empiric treatment options for a limited number of specific infections. Macrolides are one of the few available therapies for serious Campylobacter infections (particularly in children) and limited therapy for multi-drug-resistant (MDR) Salmonella and Shigella infections [7]. The emergence of ARE to macrolides has led to the development of telithromycin, which is the first clinically prescribed ketolide against macrolide-resistant strains but is rarely used because of a restricted label and liver toxicity warnings.
WHO’s high-priority critically important microbials [7] include aminoglycosides (e.g., amikacin and gentamicin), oxazolidinones (linezolid), and the tuberactinomycin capreomycin (Table 1). Aminoglycosides bind to the 30S DC region and inhibit the translocation step of elongation as well as increase the error rate [2]. Amikacin (second choice for sepsis in neonates and children) and gentamicin (first or second choice for severe pneumonia, sepsis in neonates and children, gonorrhea, and surgical prophylaxis) are listed as the WHO “access group” AB and have activity against a wide range of commonly encountered susceptible pathogens, show lower ARE potential, and are therefore recommended as essential first or second choice empiric treatment options that should be widely available, affordable, and quality assured [7]. However, aminoglycoside clinical usage has several limitations. All aminoglycosides can cause irreversible vestibular and auditory toxicity and may affect renal function [11]. Neomycin and kanamycin are limited to topical use in small amounts due to toxicity. Aminoglycosides often require intravenous administration (not well absorbed orally) and are infrequently used alone but rather used in combination with other classes of AB in order to address ARE. Aminoglycosides are the sole or a limited therapy as part of the treatment of enterococcal (ARE to aminoglycosides not uncommon) infections, MDR tuberculosis, and MDR Enterobacteriaceae. Plazomicin (approved for medical use in the United States in 2018 and sold under the brand name Zemdri), used to treat complicated urinary tract infections, is classified by the WHO as “reserve group” AB that should be reserved for the treatment of confirmed or suspected infections due to MDR pathogens and treated as “last resort” when no alternatives are available [8]. In addition to plazomicin, the “reserve group” includes the oxazolidinone linezolid whose application should also be restricted to highly specific patients and settings, when all alternatives have failed or are not suitable. In general, the “reserve group” AB usage should be monitored and reported to preserve their effectiveness in avoiding ARE emergence and spread. Further highlighting the urgency of ARE is the fact that there is a high absolute number of people affected by diseases for which either macrolides or linezolid is the sole or one of the few therapies available. Unlike macrolides that bind to NPET, oxazolidinones bind to 50S PTC A-site but also interfere with protein synthesis in a context-dependent manner, leading to unproductive binding–dissociation cycles of incoming aminoacyl-tRNAs [9][12][13]. Linezolid (the first approved oxazolidinone) has been in clinical use since 2000 and is a limited therapy for infections due to MDR Enterococcus and methicillin-resistant Staphylococcus aureus (MRSA). MRSA is a major cause of morbidity and mortality worldwide, often requiring long and costly hospital stays, and is therefore considered a serious threat by the Centers for Disease Control and Prevention (CDC). The last class of AB under the WHO’s high-priority critically important antimicrobials is ribosome subunit interface binding tuberactinomycin, which inhibits translocation and whose representative capreomycin is used in combination with other AB solely for the treatment of drug-resistant tuberculosis and other mycobacterial infections [7] (Table 1).
Moving on, phenicols (chloramphenicol and thiamphenicol), lincosamides (clindamycin and lincomycin), steroid antimicrobials (fusidic acid), streptogramins (quinupristin and dalfopristin), and tetracyclines (doxycycline) are classified as highly important antimicrobials by the WHO [7]. Like oxazolidinones, phenicols bind to PTC A-site and interfere with aminoacyl-tRNA positioning in a context-dependent manner influenced by the nature of the amino acid-forming peptide bonds in the PTC [9][12]. Therefore, phenicols cannot be considered universal inhibitors of protein synthesis, but rather modulators. Chloramphenicol is the WHO “access group” AB and one of the limited therapies for acute bacterial meningitis, typhoid, and non-typhoid fever, and respiratory infections [7][8]. Lincosamides bind to PTC A-site as well and interfere with aminoacyl-tRNA accommodation [14][15]. Clindamycin is the WHO “access group” AB used for the treatment of several bacterial infections, including strep throat, pneumonia, middle ear infections, and endocarditis. It can also be used to treat some cases of MRSA, but the WHO notes the risks of ARE [7][8]. Streptogramins include two structurally and functionally distinct subclasses: group A (SA) bind to PTC overlapping A- and P-sites, thereby inhibiting peptide bond formation; and group B (SB) bind to NPET, thereby hampering the egress of the nascent chain [10]. SA and SB act synergistically, with SA binding promoting the binding of SB. Streptogramins have been used as livestock feed additives for decades but were not approved by the Food and Drug Administration (FDA) until the introduction of quinupristin (SB)-dalfopristin (SA) (Synercid) in 1999. The clinical use of this combination therapy is limited by its intravenous administration as well as narrow spectrum of activity and is therefore reserved for hospitalized patients with MDR skin infections or with bacteremia caused by vancomycin-resistant Enterococcus faecium. Synercid is active against MRSA but ARE may result from transmission from non-human sources [7]. Fusidic acid (FA) inhibits protein synthesis by binding to elongation factor G (EF-G) on the ribosome and preventing the disassembly of the post-translocation complex; the resultant steric occlusion of the A-site by EF-G blocks the delivery of incoming aminoacyl-tRNA [16][17]. FA is the sole or limited therapy for MRSA infections; unfortunately, ARE among clinical isolates of S. aureus has increased dramatically in recent years [18]. Tetracyclines target the 30S DC A-site and inhibit the delivery of aminoacylated tRNA in the A-site. The WHO “access group” doxycycline is used to treat pneumonia, Lyme disease, cholera, typhus, and syphilis, among other infections, and is a limited therapy for infections due to Brucella, Chlamydia, and Rickettsia [7]. Finally, pleuromutilins are considered by the WHO as important antimicrobials [7] (Table 1). Pleuromutilins interact with 50S PTC A- and P-site, hindering proper positioning of tRNAs and leading to the inhibition of protein synthesis, especially at initiation codons [19]. Pleuromutilins are highly potent drugs against MDR Gram-positive and some Gram-negative bacteria [20] used in veterinary medicine and since 2007 as topical treatment in humans (retapamulin). The potential for ARE development in the clinic is predicted to be slow as confirmed by extremely low ARE rates to this class in animal infections despite the use of pleuromutilins in veterinary medicine for over 30 years [20].
The ribosome binding mode and translation inhibition mechanism of the ribosome-targeting AB classes, as well as the various ARE mechanisms (see Table 1) adopted by bacteria to overcome them, have been covered in great detail in many excellent reviews [2][3][4][18][21][22]. In short, despite the large size of the ribosome, relatively few functionally important regions (foremost PTC/NPET and DC) are targeted by the current arsenal of clinically relevant AB, which results in significant overlap between many of the binding sites. PTC-targeting AB binding sites overlap with the A-site tRNA (e.g., phenicols, lincosamides, and oxazolidinones) or span both A- and P-sites (pleuromutilins and SA). The binding sites of the macrolides and SB classes are located adjacent to the PTC within the NPET through which the growing polypeptide chain transverses during translation. Most macrolide and SB members do not inhibit peptide bond formation per se but rather prevent elongation of most nascent chains, which leads to peptidyl-tRNA drop-off and abortion of translation, resulting in imbalance in protein production. As mentioned, ARE mechanisms have been identified for virtually every ribosome-targeting AB (Table 1). Most of the prevalent ARE mechanisms are relatively well understood and their clinical significance has been recognized. These include the mutation or modification of the target sites in the ribosome to reduce or abolish AB binding. Alternatively, AB themselves can be degraded, modified, or pumped out of the cell by dedicated enzymes, thereby lowering the intracellular concentration to non-toxic levels [3][21][22].
Ribosome protection constitutes a ribosome-interacting factor-assisted target protection mechanism [18]. Unlike target alteration (AB binding site mutation or modification, e.g., ribosome methylation by specialized methyltransferases), target protection does not involve a permanent change to the target. In case of a target as conserved and functionally fine-tuned as the ribosome, permanent changes that alter the target in order to reduce or abolish AB binding are often accompanied by a fitness cost due to reduced functionality of the highly conserved centers of the ribosome [23]. Hence, there tends to be a trade-off between optimal fitness and ARE. For example, to overcome the fitness cost resulting from the methylation of 23S rRNA residue A2058 that confers resistance to macrolides, ketolides, lincosamides, and SB, the corresponding Erm methyltransferases are not expressed constitutively but are induced only in the presence of corresponding AB [24]. This inducible system allows S. aureus to survive in the presence of an AB yet still maintain optimal growth when conditions are favorable [23]. In contrast, ribosome protection results from persistent or repeated direct physical interaction between ribosome and specialized ribosome protection proteins (RPP) that does not introduce a permanent change to the ribosome in order to rescue the translation apparatus from AB inhibition. Target protection had previously not been considered a leading cause of ARE in the clinical setting; however, it has recently become evident that it can cause ARE to a vast majority of the clinically relevant translation inhibitors (Table 1). Currently, three classes of RPP have been identified: Tet-type proteins, which mediate ARE to tetracycline; Fus-type proteins, which mediate ARE to FA; and the most recent class to emerge—ABC-F proteins—which mediate resistance to diverse AB, including macrolides, oxazolidinones, phenicols, lincosamides, streptogramins, and pleuromutilins.
Due to the relative lack of major side effects and cheap cost, tetracyclines (TET) have been used extensively in the treatment of various infections in humans as well as growth promotors in agriculture, resulting in widespread ARE among clinically relevant pathogens [25]. Members of the TET class AB bind in a position overlapping with the A-site of DC in 30S and inhibit translation by interfering with the delivery of the incoming amino-acyl-tRNA by elongation factor Tu (EF-Tu) during translation elongation [26][27]. This was the first class of AB for which RPP were identified in the early 1990s [28] but the Tet-type RPP have only recently come to public attention as a significant contributor of ARE in human pathogens [18][25]. TetM and TetO are the most well known of the 15 classes of Tet-type RPP currently listed in the Comprehensive Antibiotic Resistance Database [29][30]. TetM is the most prevalent TET ARE determinant in clinical isolates of streptococci, staphylococci, as well as enterococci [18]. The TetO gene has been described in Campylobacter, Streptococcus, and Enterococcus species. Collectively, Tet-type RPP are found in a diverse range of Gram-negative and -positive pathogens, representing the major cause of ARE in the latter. Phylogenetic studies have revealed one distinct branch of TET RPP suggesting a single ancient point of origin from duplication of an elongation factor G-like gene [18]. Horizontal transmission is the main way to spread RPP-mediated ARE among bacteria (e.g., the tetM gene is found in various transposons) [25][30].
TetM and TetO catalyze the GTP-dependent release of TET from the ribosome (Figure 1A) and share a significant sequence and structural similarity with elongation factors G (EF-G) and Tu (EF-Tu) [31][32]. Cryo-electron microscopic (cryo-EM) studies indicate that Tet-type RPP have overlapping binding sites with EF-G as well as TET in the ribosome A-site [33][34], implying that ARE is mediated through direct physical displacement of the AB. Indeed, conserved proline (Pro-509) of TetM is located directly within the TET-binding site, where it interacts with the 30S 16S rRNA nucleotide C1054 [35]. As RPP becomes trapped on the ribosome in the presence of the non-hydrolyzable GTP analog, it appears that GTP hydrolysis is required for RPP dissociation rather than AB release. Conformational changes within RPP that are associated with GTP hydrolysis may not only facilitate its dissociation from the ribosome but also induce a persistent conformational change within the AB binding site. These conformational changes can likely hinder the immediate rebinding of the AB as well as promote the subsequent delivery of the aminoacyl-tRNA by EF-Tu and enable the translation to continue in the presence of TET [26][33]. Notably, RPP confer ARE to some (tetracycline, minocycline, and doxycycline) but not all TET classes of AB. For instance, tigecycline, eravacycline, and omadacycline retain translational inhibition in the presence of RPP [25]. This can be potentially attributed to the bulky side chains at the C9 position of the D-ring in these TET derivatives that mediate the AB ribosome binding mode and affinity, thereby likely rendering RPP incapable of dislodging the drugs [27][36][37]. However, the exact mechanism of RPP evasion is not fully understood and E. faecalis resistant to tigecycline due to transposon-encoded constitutively expressed TetM has been reported [38].
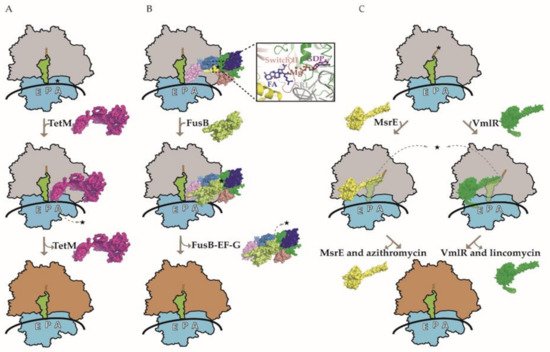
Figure 1. Three models of ribosome protection protein-mediated antibiotic resistance. (A) A model for ribosome protection against tetracycline (TET) mediated by the TetM protein. Drug-stalled ribosome with tRNA in the P-site (green) is rescued by TetM (pink), which competes with TET (shown with star) in the A-site, thereby purging it from the ribosome. The subsequent GTP hydrolysis-dependent release of TetM from the ribosome enables protein synthesis to resume. (B) A model for ribosome protection against fusidic acid (FA) mediated by the FusB protein. FA interaction with elongation factor G (EF-G) prevents its dissociation from the ribosome. FusB (lime green) interacts with the ribosome-bound EF-G, leading to its release and allowing translation to proceed in the presence of FA. The domains G, G’, II, III, IV, and V of EF-G are colored green, blue, deep salmon, yellow, and sky blue, respectively. An enlarged view of the FA binding pocket is shown, involving EF-G domains G, II, and III. EF-G switch II (residues 82–102) is colored red and the 23S ribosomal RNA sarcin-ricin loop (SRL) is colored gray. In addition, GDP and Mg2+ in the vicinity of FA are also shown. Notably, FusB does not interact with the same region of EF-G as FA and there is no evidence for direct physical displacement of the drug. (C) A model for ribosomes protection against various classes of PTC/NPET-targeting AB mediated by the ARE ABC-F proteins. Two representatives of ARE ABC-F proteins, MsrE (yellow) and VmlR (dark green), are shown to bind to the E-site of the drug-stalled ribosome. Their antibiotic resistance domain (ARD) distorts the tRNA in the P-site (green) in order to access the drug-binding site. Allosteric and/or steric interactions in PTC promote the dissociation of drugs. ATP binding may promote RPP–ribosome interaction, while ATP hydrolysis leads to the dissociation of RPP from the ribosome. Drugs corresponding to MsrE and VmlR are azithromycin and lincomycin, respectively (also shown with stars). Ribosomes with gray and orange large subunits represent translationally inactive and active complexes, respectively.
Fusidic acid (FA) is widely used as a topical treatment of staphylococcal skin infections and is one of the few remaining AB that can be used to treat MRSA orally. Unfortunately, ARE to FA among clinical isolates of MRSA and other staphylococci has increased dramatically over the last few decades [39]. Clinical ARE predominantly results from horizontal acquisition of Fus-type RPP genes that seem to originate from the duplication of accessory translation factors, which have obtained ARE activity during evolution [40][41][42].
FA acts by interfering with EF-G functioning during translation. Once translocation has occurred, EF-G dissociates from the ribosome vacating the A-site for the incoming aminoacyl-tRNA. In the presence of FA, the drug binds to EF-G on the ribosome and inhibits its release, thereby preventing disassembly of the post-translocation complex and blocking the delivery of incoming aminoacyl-tRNA [16][43]. The small two-domain metalloprotein FusB is the most studied FA RPP. FusB binds to and promotes the dissociation of FA-trapped EF-G from the ribosome, allowing translation and/or ribosome recycling to resume in the presence of the drug [42][44][45] (Figure 1B). In contrast to other RPP, Fus proteins do not interact with its target in the proximity of the bound AB. More specifically, Fus protein makes contacts with domain IV and V of ribosome-bound EF-G, whereas FA binds to a pocket between domains G and III [16][40]. There is no evidence for direct physical displacement of AB by FusB. Instead, Fus-type RPP negate the EF-G ribosome tethering effect of FA by inducing conformational changes in EF-G domains IV and V as well as the dynamics of domain III, enabling EF-G dissociation from the ribosome [40]. In other words, Fus-type protein-mediated modulation of EF-G function can overcome FA inhibition, resulting in ARE. FA is likely to dissociate from free EF-G due to low affinity.
ATP-binding cassette subfamily F (ABC-F) proteins first gained attention as ARE-mediating RPP about 5 years ago when Sharkey et al. showed that purified ABC-F proteins (S. aureus VgaA and E. faecalis LsaA) are capable of displacing AB (SA virginiamycin M and lincomycin, respectively) from the ribosome and rescuing translation in vitro [46]. These experiments provided the first direct evidence to the hypothesis that ABC-F proteins mediate ARE through target protection [47]. Since then, it has become apparent that ABC-F represents a widespread family of proteins that collectively provide ARE to a broader range of clinically important AB classes than any other group of ARE proteins. Indeed, ABC-F members constitute a major source of clinically significant ARE to almost all classes of PTC/NPET-targeting AB (including macrolides, oxazolidinones, phenicols, lincosamides, streptogramins A and B, as well as pleuromutilins) [48][49][50][51] (Table 1).
In-depth study of ABC-F subfamily members across all species with sequenced genomes revealed that, unlike Tet- and Fus-type RPP, known ARE ABC-F proteins are not confined to a distinct phylogenetic lineage [18][51]. Instead, numerous phylogenetic lineages (ARE 1-8 in Table 2) exist suggesting that ARE has arisen on several occasions among ABC-F of unknown function (e.g., potential translation factors). ARE emergence notably benefits the cell and is likely to be retained during evolution, especially if it comes without the loss of cellular fitness. As yet uncharacterized bacterial ABC-F subfamily members cluster with known groups of ARE ABC-F proteins, it seems likely that additional members of the ABC-F mediating clinically relevant ARE remain to be discovered [51]. Furthermore, non-ARE ABC-F protein can readily evolve to gain ARE function given sufficient pressure from mis- and overuse of AB [52].
Table 2. List of ARE ABC-F proteins in pathogens and antibiotic producers with the respective hosts and resistance profiles.
| Phylogenetic Lineage | ARE ABC-F in Pathogens and AB Producers | Species | Resistance Phenotype | Drug Binding Site |
|---|---|---|---|---|
| ARE 1 | MsrA | Staphylococcus aureus, Staphylococcus epidermis | macrolides, ketolides, and group B streptogramins (MKSB) | NPET |
| MsrC | Enterococcus faecium | |||
| MsrD | Streptococcus pyogenes, Streptococcus pneumoniae | |||
| MsrE | Pasteurella multocida, Pseudomonas aeruginosa, Escherichia coli | |||
| VgaA | Enterococcus faecalis, Staphylococcus aureus, Staphylococcus haemolyticus | pleuromutilins, lincosamides, and group A streptogramins (PLSA) | PTC A-site overlapping with P-site and NPET | |
| VgaB | Staphylococcus aureus | |||
| VgaC | Staphylococcus aureus | |||
| VgaD | Enterococcus faecium | |||
| VgaE | Staphylococcus aureus | |||
| ARE 2 | VmlR | Bacillus subtilis | pleuromutilins, lincosamides, and group A streptogramins (PLSA) | PTC A-site overlapping with P-site and NPET |
| ARE 3 | EatA | Enterococcus faecium | pleuromutilins, lincosamides, and group A streptogramins (PLSA) | PTC A-site overlapping with P-site and NPET |
| LsaA | Enterococcus faecalis | |||
| LsaB | Staphylococcus sciuri | |||
| LsaC | Streptococcus agalactiae | |||
| LsaE | Staphylococcus aureus | |||
| ARE 4 | CarA | Streptomyces termotolerans | specific to AB produced by each species | PTC A-site overlapping with NPET |
| OleB | Streptomyces antibioticus | |||
| SrmB | Streptomyces ambofaciens | |||
| TlrC | Streptomyces fradiae | |||
| ARE 5 | LmrC | Streptomyces lincolnensis | specific to AB produced by each species | PTC A-site overlapping with P-site |
| VarM | Streptomyces virginiae | |||
| ARE 6 | SalA | Staphylococcus sciuri | pleuromutilins, lincosamides, and group A streptogramins (PLSA) | PTC A-site overlapping with P-site and NPET |
| ARE 7 | OptrA | Enterococcus faecalis | oxazolidinones and phenicols (PhO) | PTC (A-site) |
| ARE 8 | PoxtA | Staphylococcus aureus | oxazolidinones and phenicols (Pho) | PTC (A-site) |
Extensive phylogenetic studies have determined that ARE ABC-F members are widespread in the chromosomes of Gram-positive and, to a lesser extent, Gram-negative bacteria, as well as in mobile genetic elements of many clinically isolated pathogens [48][51][52][53][54][55].
These include the ESKAPE species (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) that contribute to a substantial portion of hospital-acquired MDR infections. Notably, ABC-F (e.g., the optrA and poxtA genes) mediate ARE against the clinically crucial AB linezolid (Table 1), which is used in the treatment of MRSA nosocomial strains and vancomycin-resistant Enterococcus faecium [53]. Moreover, the optrA gene, whose presence has also been reported in other staphylococci, is to date the only known horizontally transmissible determinant capable of conferring ARE to tedizolid, a second-generation oxazolidinone approved by the FDA in 2014 [54]. Pseudomonas aeruginosa (P. aeruginosa) is a common Gram-negative pathogen whose infections have become increasingly severe due to the spread of MDR to various AB, especially β-lactams, aminoglycosides, quinolones, and sulfonamides, resulting in very limited treatment options [55]. Indeed, carbapenem-resistant P. aureus ranks 2nd (following carbapenem-resistant A. baumannii; the infamous MRSA ranks 12th) in the final ranking of the WHO’s 2017 “Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis” [56]. Macrolides have been used to treat MDR P. aeruginosa infections; however, due to increasing applications in clinical practice, MsrE-mediated ARE to macrolides has started to spread worldwide [57][58]. MsrD protein plays a predominant role in conferring macrolide ARE to Streptococcus pneumoniae and Streptococcus pyogenes isolates in various parts of the world, including the US and the UK [59]. In staphylococci, msr-type RPP (particularly MsrA) are responsible for ARE in up to 30% of the strains exhibiting the MKSB (ARE to macrolides, ketolides, and SB) phenotype [48]. ABC-F is also a major contributor to pleuromutilin ARE in staphylococci as the vga genes account for all instances of ARE to retapamulin in the nearly 6000 S. aureus isolates tested [60]. While altogether the incidence of Vga-mediated pleuromutilin ARE in human S. aureus isolates is considerably lower than in isolates from animals (where it has spread due to extensive use of pleuromutilins in food production and agriculture), this is very likely to change in the future in response to the recent (2019 in the US and 2020 in Europe) approval of the first systemic pleuromutilin lefamulin (sold under the brand name Xenleta) in human medicine [61][62]. ABC-F genes can disseminate easily from strain to strain via MDR conferring plasmids, and many examples of horizontal gene transfer have been reported [63]. Consequently, ABC-F family proteins can be an important source of ARE in “superbugs.”
No individual ABC-F protein confers ARE to all of the PTC/NPET-targeting AB. Three distinct profiles can be distinguished in clinical isolates: the MKSB phenotype conferring ARE to macrolides, ketolides, and SB (e.g., Msr proteins); the PLSA phenotype conferring ARE to pleuromutilins, lincosamides, and SA (multiple variant proteins arising from distinct bacterial lineages, e.g., Vga and Lsa proteins); and the Pho phenotype conferring ARE to phenicols and oxazolidinones (e.g., OptrA and PoxtA) [18][51] (Table 2). ARE 4 and ARE 5 phylogenetic lineage members provide self-protection against various classes of drugs in AB-producing bacteria such as Streptomyces. Notably, cross-resistance mediated by individual ARE ABC-F proteins to different AB correlates with spatial overlap of AB binding sites (Table 2). This phenomenon has become better understood in recent years following the structural and functional characterization of the ribosome protection mechanism of several members of the ARE ABC-F proteins.
ABC-F proteins consist of two tandem nucleotide binding domains (NBD) in a single polypeptide chain connected by a 60–100-residue linker (known as the ARD for antibiotic resistance domain or the PtIM for P-site tRNA interaction motif) that is the defining feature of the ARE ABC-F family [46][48][51]. The ARD forms an α-helical hairpin containing an inter-helical loop of variable length [49][64][65]. The ARD inter-helical loop differs considerably in length and sequence among the ARE ABC-F proteins, and alterations within this sequence can alter the AB specificity [18][46][49]. ARE ABC-F proteins may include an “arm” subdomain within the NBD1 as well as an additional C-terminal extension (CTE). The first structural insight into how ABC-F proteins confer ARE came from the cryo-EM structure of P. aeruginosa macrolide resistance protein MsrE bound to the T. thermophilus ribosome with a cognate deacylated tRNA in the P-site [64]. MsrE protein was trapped on the ribosome by using a non-hydrolysable ATP homolog. Shortly thereafter, the cryo-EM structure of Bacillus subtilis (B. subtilis) lincomycin and SA resistance-associated ATPase-deficient VmlR protein in complex with the ErmDL-stalled B. subtilis ribosome was reported [65]. Several recent reviews have compared the two structures in detail [49][50][52] and a common theme of the RPP mechanism is emerging (Figure 1C). Namely, the substrate is the AB-stalled ribosome and the RPP bind in the vacant exit (E) site in the ATP form adopting the closed conformation with the ARD reaching deeper into the ribosome towards the PTC/NPET region. In both the MsrE and VmlR structures, the P-tRNA interacts with the RPP, resulting in its notable shift toward the E-site, while the acceptor stem is shifted away from the PTC toward a site usually occupied by the acceptor stem of the fully accommodated A-tRNA [64][65]. RPP likely stabilizes the P-tRNA to prevent its drop-off. These conformational changes of P-tRNA allow the ARD to reach PTC in the VmlR structure as well as the adjacent NPET region in the MsrE structure. It should be noted that there is a correlation between the length of the inter-helical loop, its positioning in the ribosome, and the corresponding AB binding site. VmlR has a shorter loop and confers ARE to lincosamides, SA, and pleuromutilins, which target PTC A-site overlapping with P-site and NPET, while MsrE has a notably longer loop that projects deeper into NPET where the macrolides bind. MsrE ARD loop leucine residue (Leu-242) comes within 1.8 Å and could clash with the ribosome-bound macrolides and SB [64]. Substituting this leucine with alanine leads to a near-complete loss of MsrE’s ability to mediate azithromycin (AZM) ARE, which also suggests a steric component to drug release [64]. In addition, conformational changes in the PTC region and a slight widening of the NPET around the macrolide binding site are observed in the MsrE-ribosome structure. Similar conformational changes in the PTC region are observed in the VmlR-ribosome structure; however, direct steric interference between the RPP and AB seems not critical based on the cryo-EM structure as well as mutagenesis studies of the VmlR phenylalanine (Phe-237) residue that comes closest to the bound AB [65]. Curiously, neither MsrE nor VmlR confer resistance to oxazolidinones and phenicols, even though their binding sites on the ribosome overlap. Furthermore, it is not clear how OptrA and PoxtA manage to dislodge oxazolidinones and phenicols from the ribosome as these proteins have a relatively short ARD that is not expected to reach into the PTC where the corresponding AB bind [18].
The ATPase activity of ABC-F is critical for ARE [49][64][66]. ATP hydrolysis does not seem to be required for AB release per se; instead it likely drives the two NBD domains apart triggering the release of RPP from the ribosome so that translation can resume. Murina et al. have shown that VgaA can hydrolyze other NTPs as well and operates as a molecular machine requiring NTP hydrolysis (not just NTP binding) for ARE [66]. Persisting allosteric changes in the ribosome (as is the case following Tet-mediated TET release discussed previously), tRNA (re)-accommodation into PTC A- and P-sites or nascent chain settlement into NPET (i.e., following macrolide release) likely block AB rebinding. Furthermore, AB displacement activity could be coupled to functional partners that degrade or pump AB out of the cell. Although continual dynamic displacement of AB driven by RPP ATPase activity might be required to “plow through” the stalling-prone sequences of certain nascent chains in the presence of the context-dependent translation inhibitors (macrolide, oxazolidinones, and phenicols), RPP rebinding is likely hindered by deacylated tRNA progression into the E-site when AB no longer poses an issue.
Taken together, comparison of the RPP from different ABC-F phylogenetic lineages and with different ARE profiles suggests a common yet adaptable mechanism of ARE to translation elongation inhibitors, which trap the ribosome with the tRNA in the P-site, resulting in slow or stalled translation. AB displacement is achieved by a combination of direct interactions of RPP with the drug as well as by ARD loop-mediated allosteric relay of changes to the PTC and/or NPET in the vicinity of the AB binding site by altering the orientation of rRNA residues involved in AB binding.





