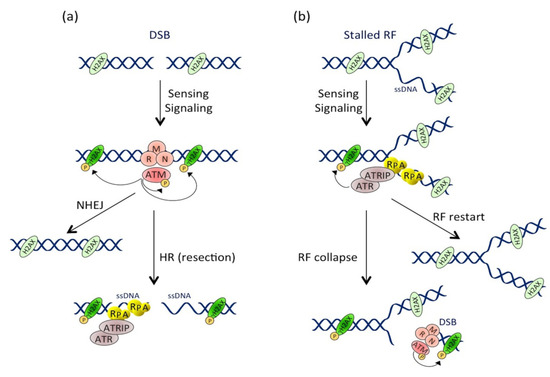
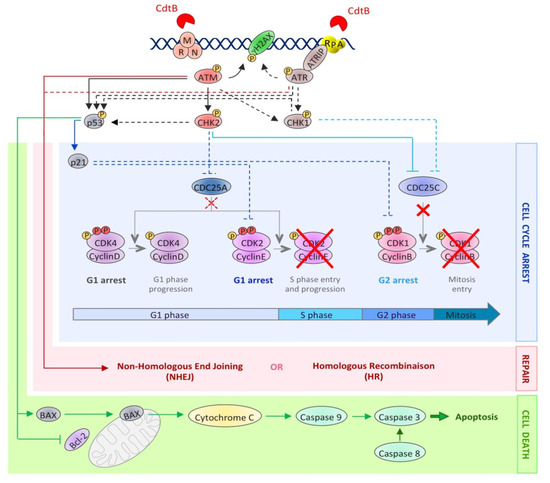
The DNA damage-related activation of checkpoints is divided into three stages: (i) the recognition of DNA damage by sensor proteins (MRN and Ku complexes, RPA), which rapidly activate specific phosphatidylinositol 3-kinase-related protein kinases (ATM, ATR, DNA-PK)
[55]; (ii) the signal amplification by transducing proteins (CHK1, CHK2); which (iii) activates an appropriate cellular response by effectors proteins (p53, CDC25,
etc.). This cell response initiates the cell cycle regulation, the activation of DNA repair pathways and, in some cases, cell death pathways
[56].
Two key signaling pathways are activated in response to DNA damage: the ATM-CHK2 and the ATR-CHK1 pathways. The ATM-CHK2 pathway is activated with the response to DSB-inducing agents. Following the generation of a DSB, the MRN complex, consisting of MRE11, RAD50 and NBS1, recruits the ATM kinase to the site of injury
[57]. Once at the DSB site, ATM is activated by autophosphorylation and phosphorylates hundreds of substrates, including CHK2 and p53. Meanwhile, ATM phosphorylates the H2AX histone (then called γH2AX), several megabases around the DSB site
[58], allowing signal amplification. The activated CHK2 phosphorylates various substrates, including p53 and the CDC25 phosphatases family. By contrast, the ATR-CHK1 pathway is activated by the accumulation of single-stranded DNA (ssDNA), particularly during the stalling of replication forks (RFs). Indeed, when replication is blocked by DNA lesions (SSB, DSB, inter-strand and intra-strand crosslinks, base modifications or adducts), DNA polymerase is uncoupled from the replicative helicase, which continues to unwind the DNA and, thus, generates ssDNA. ssDNA is recognized by the RPA protein complex, which protects and stabilizes it, and the accumulation of RPA-coated ssDNA at stalled RFs induces the recruitment of the ATR/ATRIP complex
[59]. ATR then phosphorylates the CHK1 transducing protein
[60]. ATR can, like ATM, phosphorylate H2AX
[61], p53 and many cell cycle regulators (such as CDC25A, CDC25C and Wee1). In conclusion, whatever the activated pathway, the ATR-CHK1 or ATM-CHK2 activation will lead to major protein phosphorylation, involved in various cellular processes, including cell cycle regulation, DNA repair and programmed cell death
[62]. However, it has to be underlined that many crosstalk exist between the ATM and ATR pathways (reviewed in
[63]). Indeed, although CHK2 is the ATM primary target, ATM can also phosphorylate CHK1. In addition, processing of DSBs during the homologous recombination pathway (HR) generates stretches of ssDNA, leading to the ATR pathway activation
[64]. Conversely, prolonged replicative stress can provoke RF collapse
[65], resulting in DSB formation and ATM-CHK2 pathway activation (). In summary, according to the type of DNA injury, the ATM and ATR pathways can be specifically or sequentially activated, resulting in the cell cycle arrest, the activation of the DNA repair machinery and, potentially, in cell death.
 +1 point
+1 point