ATM is among of the most critical initiators and coordinators of the DNA-damage response. ATM canonical and non-canonical signaling pathways involve hundreds of downstream targets that control many important cellular processes such as DNA damage repair, apoptosis, cell cycle arrest, metabolism, proliferation, oxidative sensing, among others. Of note, ATM is often considered a major tumor suppressor because of its ability to induce apoptosis and cell cycle arrest. However, in some advanced stage tumor cells, ATM signaling is increased and confers remarkable advantages for cancer cell survival, resistance to radiation and chemotherapy, biosynthesis, proliferation, and metastasis.
1. Introduction
During the past three decades, the study of ATM (ataxia telangiectasia mutated) has played a central role in advancing our understanding of the mammalian DNA damage response, cancer initiation and progression as well as redox signaling pathways. Numerous recent publications additionally reported the varied roles and influences of ATM in multiple cellular processes, for instance, growth, metabolism, energy production, oxidative homeostasis, chromatin remodeling, and genomic integrity, which are all important processes in tumor development and progression. Although ATM should be considered as a tumor suppressor because its mutations often lead to cancer, there are mounting evidence showing that ATM’s functions and signaling pathways may also contribute to cancer cells’ resistance to radiation, chemotherapy, and even assist tumor progression in some scenarios.
2. The Structure and Domain Mapping of ATM
The ATM gene consists of 66 exons and is located on chromosome 11 (11q22-23)
[1]. In its active monomeric form, ATM has a large size of 370 kDa. ATM is an important member of the PI3K-related protein kinase family (PIKK)
[2]. Other members of this family include DNA-dependent protein kinase catalytic subunit (DNA-PKcs), ataxia-telangiectasia and Rad3-related (ATR), mammalian target of rapamycin (mTOR), SMG-1, TRRAP, among others
[3][4][5]. Similar to these members, ATM possesses a kinase domain whose structure shares significant homology to that of the phosphatidyl inositol 3 kinase (PI3K). Thanks to this domain, ATM is an active serine/threonine kinase. Besides, ATM also has a FAT (FRAP (FK506-binding protein 12-rapamycin-associated protein (mTOR)), ATM, TRAPP (Transformation/transcription domain-associated protein)) domain and a FATC domain that is located on its C-terminal
[6]. In 2006, Jiang et al. additionally showed that the FATC domain was essential for ATM to interact with its partners for activation and control of its kinase activity
[7].
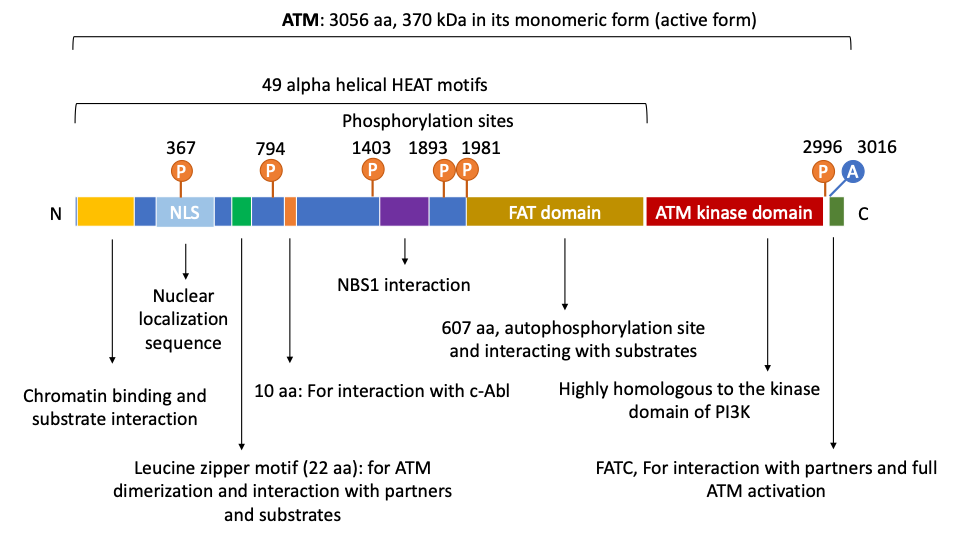
The N-terminus of ATM contains multiple α helical HEAT repeat motifs and a region that appear to be critical for ATM interactions with other proteins and DNA by serving as scaffolds. It has been shown that HEAT motifs are necessary for ATM to interact with and recruit a number of proteins to the DNA lesion sites for DNA damage repair
[8][9][10][11]. In fact, ATMIN (ATM interacting protein), NBS1 (a component of the MRN complex), and NKX3.1 (a prostate tumor suppressor) have been found to bind to ATM’s HEAT motifs
[12][13][14][15]. In 2004, Lavin et al., also reported the presence of other motifs, a proline-rich area for c-Abl binding, and a leucine zipper on ATM
[16][17] (Figure 1).
Figure 1. Human ATM’s domain map. ATM is a large gene with 66 exons and consists of multiple domains that allow its protein to directly interact with numerous regulators, partners, and downstream targets. In its N terminus, ATM contains a Nuclear Localization Signal that enables its nuclear translocation. ATM also uses its N-terminus for interacting with chromatin, substrates and partners, for instance, p53, LKB1, NBS1, among others. The putative leucine zipper motif on ATM N-terminus is documented to be essential for its dimerization and interaction with other partners or substrates. On its C-terminus, ATM has a FAT (FRAP, ATM and TRRAP proteins) domain which contains autophosphorylation sites and is critical for substrate binding. As a kinase, ATM has a serine/threonine kinase domain that is highly homologous to that of the PI3K. At the end of its C-terminus is the FATC domain that is essential for ATM full activation and interactions with partners. Of note, ATM contains multiple important autophosphorylation sites that can substantially affect its functions. For instance, Ser367, Ser1893, Ser1981, and Ser2996 are auto-phosphorylated after irradiation. Thr1885 is not induced by irradiation. In human cells, autophosphorylation on three residues Ser367, Ser1893, Ser1981, and acetylation on Lys3016 have been shown to be important for ATM activation. Aurora B phosphorylates ATM at Ser1403 during mitosis. Importantly, ATM also has 49 α helical HEAT motifs whose name is derived from huntingtin, elongation factor 3, the A subunit of protein phosphatase 2A, and target of rapamycin 1 (TOR1). These motifs serve as scaffolds and are critical for ATM interactions with proteins and DNA.
3. ATM’s role in DNA Damage Response and Ataxia Telangiectasia
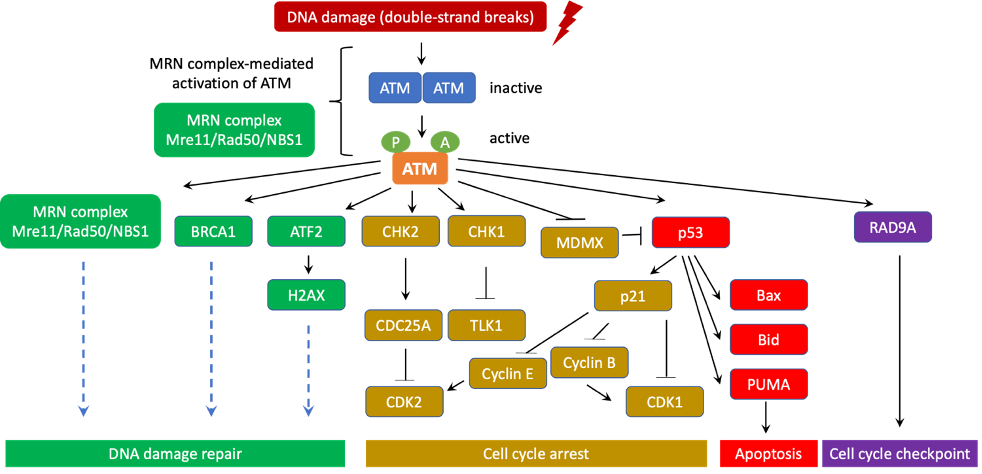
ATM is an important initiator of the DNA-damage response in mammalian cells via the Mre11/Rad50/Nbs1 (MRN) complex at the DNA lesion sites
[18][19][20][21]. ATM activation occurs after its direct binding to the MRN complex, leading to its conformational change from a resting homodimer or polymer state to an active monomer form
[22]. Active ATM then uses its kinase activity to phosphorylate a number of downstream targets that are essential for DNA damage repair, apoptosis, cell cycle arrest, and cell-cycle checkpoints
[21][23][24][25]. Therefore, ATM is often considered a major tumor suppressor (Figure 2).
Figure 2. ATM canonical signaling pathway in DNA damage repair, cell cycle arrest and apoptosis. In normal situations, ATM are often inert and form homodimers or polymers. However, when DNA damage occurs, ATM is quickly activated and disassociates into highly active monomers. ATM initiates the DNA-damage response through co-signaling with the MRN (Mre11/Rad50/NBS1) complex at the DNA lesion sites. In fact, direct interaction with the MRN complex induces ATM to phosphorylate a number of downstream targets that are essential for DNA damage repair, cell cycle checkpoint, cell cycle arrest, and apoptosis. During this process, ATM undergoes a series of autophosphorylation on Ser367, Ser1893, Ser1981, and Ser2996 as well as acetylation on Lys3016. Activated ATM is recruited to the DNA damage sites for starting the DNA-damage response. ATM activates BRCA1 and ATF2 to promote cascades of DNA damage repair signaling pathways that involve hundreds of sensors, transducers, and effectors. In addition, ATM also turns on and stabilizes p53 via direct phosphorylation. ATM additionally phosphorylates MDMX to induce its degradation, thereby further stabilizing p53. To its turn, p53 translocates into the nucleus to transactivate a series of its downstream tumor suppressor target genes. For instance, p53 enhances the expression of p21, a potent cyclin-dependent kinase inhibitor. p21 associates with cyclin E, CDK2, CDK4/6 and induces G1/S and G2/M cell cycle arrest, which is critical to prevent unrepaired DNA mutations from passing into daughter cells. p53 also directly transactivates Bid, Bax, PUMA to induce apoptosis when DNA damage is too severe for effective repair. This programmed cell death is a major mechanism of tumor suppression. Moreover, ATM activates RAD9A to further promote cell cycle checkpoints. CHK1 and CHK2 are additionally induced by ATM through phosphorylation. CHK1 subsequently inhibits TLK1 while CHK2 turns on the cell cycle inhibitor CDC25A to block CDK2. As a result, cell cycle progression is temporarily halted to enable DNA damage repair. Thus, thanks to its central role in coordinating DNA damage repair, cell cycle arrest, cell cycle checkpoint, and apoptosis, ATM is frequently considered a major tumor suppressor whose mutations often lead to significant increase in risk of cancer.
Mutations of the
ATM gene leads to deficiencies in the DNA-damage response, which results in the development of ataxia telangiectasia, a rare hereditary autosomal recessive disorder with a 1 in 40,000 to 300,000 frequency in Caucasians
[26]. This condition is characterized by radiosensitivity, cancer predisposition, immunodeficiency (frequent infections), telangiectasias of the conjunctivae, chreoathetosis, and progressive neurodegeneration (cerebellar ataxia, oculomotor apraxia)
[27][28][29][30][31][32]. In 1988, Gatti et al. used genetic linkage and found the location of the gene responsible for ataxia-telangiectasia on chromosome 11q
[30]. In 1995, Savitsky et al. applied positional cloning and identified the gene
ATM (ataxia-telangiectasia mutated)
[1]. Of note, most of ataxia-telangiectasia patients carry hereditary heterozygote or homozygote
ATM mutations from each parent. More than 300
ATM mutations have been identified and most of them involve insertions, deletions and base substitutions that result in abnormal mRNA splicing or premature termination codons.
ATM mutations prevalence was estimated to vary from 0.5% to 1% in Caucasian populations, according to Swift et al., 1986 and Renwick et al., 2006
[26][33].
 +1 point
+1 point