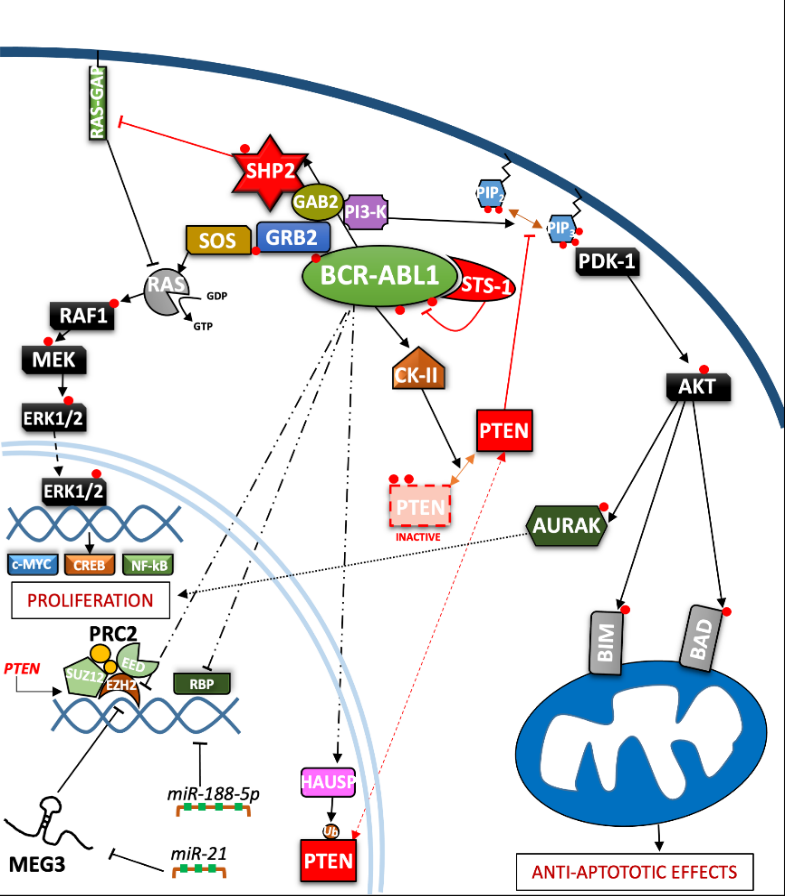
In the last three decades, the role of the BCR-ABL1 protein has been extensively analyzed and we have summarized the main pathways involved in Figure 2. One of the major signaling cascades that is altered involves the RAS mitogen-activated protein kinase family (MAPKs)
[19], such as ERK1/2, MEK, JNK, and p38 families, which lead to insensitivity to growth factor stimuli and regulates the proliferative fate of cells
[20][21]. In the active conformation, BCR-ABL1 possesses several tyrosine sites, such as Y245 in the SH2-kinase linker and Y412 in the activation loop, which are known to regulate function/activity (Figure 1). The human
UBASH3B gene encodes for STS-1 phosphatase (suppressor of T-cell receptor signaling 1), the high expression of which has been correlated with the p190
BCR-ABL1 form in acute lymphoblastic leukemia (ALL) patients’ samples
[22], but is usually considered a BCR-ABL1 interactor
[23][24]. In particular, other than being involved in signaling by other kinases, such as PDGFR, ZAP-70, and SYK, it can bind to the SH2–SH3 compartment of BCR-ABL1, producing strong dephosphorylation in the tyrosine sites of the ABL-portion, thus limiting its kinase activity. Remarkably, a CML-like disease mouse model with
Sts-1/Sts-2 double knockout exacerbated the classical CML parameters and reduced mice survival, providing evidence supporting the leukemogenic role of Sts-1
[25]. The SH2-domain containing protein growth factor receptor bound protein-2 (GRB2) recognizes another fundamental Y177 binding site on the BCR domain which synergistically promotes CML, supporting RAS activation through Son of Sevenless (SOS) a guanine nucleotide exchange factor protein, and the scaffold adapter GAB2 (GRB2-associated binding protein 2)
[26]. BCR-ABL1
+ cells harboring a Y177F or SH2-domain mutation on GRB2 have exhibited less leukemogenic transformation and reduced proliferation by MAPK pathway disruption
[27][28]. A recent study has also suggested that Y177 BCR-ABL1 phosphorylation is also regulated by ERα36-expression, an alternatively spliced variant of estrogen receptor α66 (ERα66), which is abnormally localized in the cytoplasm and cell membrane of BCR-ABL1
+ cells. Synthetic ERα36 inhibitors prevented GRB2 from binding to BCR-ABL1, generation reduction in the downstream RAS/MAPKs pathway and cell proliferation impairment
[29]. The major SHP2-binding protein in hematopoietic cells, GAB2, is part of the GRB2/GAB2 complex recruited on phosphorylated Y177. GAB2 is essential for myeloid and lymphoid leukemogenesis induced by BCR-ABL1, as demonstrated by the failure to develop a CML-like disease in a mouse model transplanted with BCR-ABL1 in
GAB2−/− marrow cells. Its tyrosine phosphorylation in BCR-ABL1
+ cells generates a docking site for signal cascade proteins, such as PI3K sub-unit p85α and SHP2
[30]; moreover, it induces the signal events, together with RAS activation, that lead to an increase of ERK function
[27]. Downstream proliferative effects of the MAPKs pathway include transcription factors (TFs) such as NF-κB, CREB, ETS-1, AP-1, and cMYC, leading to cell cycle progression (involving CDKs) and anti-apoptotic mechanisms (involving BCL-2)
[31]. SHP2 (
PTPN11) plays a critical role in cell development due to its ability to support RAS/MAPK signaling, in response to numerous growth factors
[32][33]. In the hematopoietic compartment, it is considered an oncogene as point mutations in its N-terminal SH2 inhibitory domain trigger the development of leukemia in different lineages
[34]. An early study performed in CML CD34
+ cells reported that the cytokine-independent colony formation capacity was altered by
PTPN11-knockdown
[35]. In particular, both mRNA and phosphoprotein levels appear to be greater in myeloid leukemia cells, addressing its presence/activity with a more hyper-proliferative phenotype
[36]. Further evidence has confirmed SHP2 is required to initiate and maintain BCR-ABL1-mediated transformation, as GAB2 mutation in SH2 domain cannot bind SHP2 and, as a result, reduce myeloid and lymphoid leukemic burden. In line with this observation, various studies have described p-ERK activity reduction in BCR-ABL1 expressing myeloid cells lacking SHP2 phosphatase, thus emphasizing its role as a positive regulator of the RAS/MEK/ERK1-2 pathway in BCR-ABL1 signaling
[27][37][38][39]. Speculation on reduced cell viability in this case may be traced back to RAS activation which occurs through SOS, but which also requires active SHP2 phosphatase. SHP2 can dephosphorylate and inactivate the p120 RAS-GAP protein (RAS GTPase-activating protein) by blocking its antagonistic action on RAS
[33][40]. Although there are also implications for p-AKT-reduction, it seems to be mostly associated with GAB2 adapter protein impairment. On the other hand, Y177-GAB2 interaction is required to enforce another key player in leukemic transformation: Phosphoinositide-3 kinase (PI3K)
[27] (see Figure 2).
The PI3K/AKT signal pathway plays a crucial role in a great variety of cancers, due to its strong relation with membrane tyrosine kinase receptors which are activated by multiple extracellular stimuli (cytokines, growth factors, etc.)
[41]. Reportedly, the PI3K lipid kinase consists of p85 (regulatory) and p110 (catalytic) sub-units in a heterodimeric structure that is involved in inositol lipids (PtdIns(3,4)P2) phosphorylation, usually at the inner leaflet of the cytoplasmic membrane, initiating and controlling multiple cellular functions
[42]. Specifically, PtdIns(3,4,5)P3 provides an anchor point for pleckstrin homology (PH) domain-containing proteins, such as activated protein kinase (AKT) and phosphoinositide-dependent protein kinase-1 (PDK-1). AKT is a serine/threonine kinase, belonging to the AGC family kinases, which is expressed in two isoforms in hematopoietic stem cells
[43]. Activated p-AKT following by BCR-ABL1-activated PI3K, is able to inhibit the apoptotic process and support cell proliferation
[44][45]. Skorski et al. reported the first direct data on PI3K (both sub-units) involvement in CML cell transformation
[46], demonstrating how its downstream effector, AKT, was a critical growth regulatory switch in BCR-ABL1-expressing cells
[47]. BCR-ABL1 can directly up-regulate AURK-A and AURK-B (serine/threonine kinases belonging to the Aurora family) through (at least in part) AKT, emphasizing a further pathway for cell, proliferation
[48]. The PI3K pathway is constitutively activated in CML progenitor cells, as has been demonstrated
by the elevated PtdIns(3,4,5)P3 levels found in CML progenitor cells, compared to their normal counterpart. Normal levels of PtdIns(3,4,5)P3 were restored, along with their capacity to respond near-normally to cytokine stimulation, by Imatinib treatment [49]. Many studies on this signaling pathway have been deepened in CML biology, thanks to the development of specific inhibitors to curb a wide range of effectors acting on this leukemia [50][51]. Notoriously, phosphatase and tensin homologue (PTEN) was the first tumor suppressor gene found to have a double specific phosphatase activity, acting on serine/threonine and tyrosine residues and the major antagonist of PI3K signaling, operating as a tumor suppressor in numerous solid neoplasms and leukemia [52][53]. By exploring the function of PTEN in BCR-ABL–expressing Ba/F3 cells, it has been revealed how BCR-ABL1 may induce PTEN phosphatase downregulation and a reduced p53 protein expression, regaining expression of both targets after TKI treatment [54][55]. An explanation was found in Pten gene promoter, where p53 can bind and promote its expression. Additionally, BCR-ABL1-expressing LSK cells sorted from CML mice, confirm a reduction of Pten mRNA, which also correlates with the downregulation of p53. Therefore, reduced disease progression related to PTEN expression could involve the reduction of CML cell proliferation through cell cycle arrest [54]. The nuclear-cytoplasmic shuttling enables PTEN to play its proper tumor suppressive function [56]. The mono-ubiquitinated PTEN form is predominantly nuclear, where it has been shown to play a tumor-suppressive role through a proliferation control mechanism. Herpesvirus ubiquitin-specific protease (HAUSP) is a critical modulator of protein ubiquitination, possibly regulating PTEN cytosolic partitioning [57]. In CML, BCR-ABL1 might phosphorylate HAUSP, triggering PTEN nuclear exclusion and causing proliferative advantages. Therefore, HAUSP together with nuclear PTEN depict a pivotal pathway in BCR-ABL1-induced proliferation [58]. In this way, Morotti et al. showed that PTEN-tail phosphorylation, mediated by BCR-ABL1-activated casein kinase II (CKII), caused a reduction of PTEN phosphatase activity, thus uncovering another inhibition mechanism present in CML [59]. Many different mechanisms might regulate PTEN expression in leukemic cells, including epigenetic shutdown, genomic loss, transcriptional repression, post-transcriptional regulation by lncRNA or microRNAs, etc. Polycomb repressive complex 2 (PRC2) contains two methyltransferase sub-units called enhancers of zeste 2 polycomb repressive complex subunit 1 and 2 (EZH1 and EZH2) that can modulate chromatin conformation by adding methyl groups on histone 3 (H3K27me2/3) [60]. EZH2 expression has been found to be upregulated in all three-phases of the disease in CML LSCs and, in particular, compared to other PRC2 components, it appears to be BCR-ABL1-kinase activity-dependent; meanwhile, EZH1 did not follow this trend, but was found to be downregulated [61][62]. Proliferation, self-renewal and viability of CML cells were drastically impeded by pharmacological interference with EZH2 activity (GSK126 inhibitor), or by reducing its expression through shRNA or in Ezh2−/− CML mice, resulting in increased survival in retroviral BCR-ABL1-transduced mouse models [61][63]. In addition, EZH2 knockdown decreased H3K27me3 levels on PTEN gene promoter and resulted in a significant increase in PTEN mRNA and protein expression in three human Ph+ cell lines, as well as in LSK (Lin−/Sca-1+/c-Kit+) cells from CML mice. Zhou et al. demonstrated the EZH2 inhibition-mediated beneficial effects on leukemia cells and prolonged survival of CML mice were compromised by the concurrent transduction of shRNA targeting PTEN [63]. Modification of PTEN expression might occur through maternally expressed gene 3 (MEG3), a long non-coding RNA (lncRNAs) associated with many cancers which has already been shown to regulate IM resistance in CML [64]. Lower MEG3 levels have been found in advanced stages of leukemia while, to the contrary, miR-21 was found to be over-expressed which was able to interact with MEG3 by decreasing its expression. The data indicated that MEG3 binding could modify the expression of MDM2 and EZH2 mRNA levels, producing DNA (cytosine-5)-methyltransferase 1 (DNMT1) protein upregulation and PTEN protein downregulation. Therefore, a reduced expression of miR-21 blocked the proliferation and promoted apoptosis of CML cells
[65]. The upregulation of miR-188-5p represents another post-transcriptional regulatory mechanism for PTEN expression exhibited by CML cells. Indeed, miR-188-5p may directly target PTEN 3′-UTR, thus repressing it. Zi-Yuan Nie et al. demonstrated how the flavonoid Morin, isolated from
Moraceae, might inhibit proliferation and induce apoptosis by repressing miR-188-5p expression, leading to PTEN/AKT pathway inhibition in CML cells both in the K562 cell line and mouse xenograft models
[44]. Yin et al. obtained relevant data on other epigenetic modifications produced by the RBP2 protein. In particular, the BCR-ABL1/RBP2/PTEN pathway represents a feedback loop which is thought to increase proliferation, leading to blast phase (BP) transition. BCR-ABL1 activity directly inhibits directly RBP2 protein expression, resulting in the inhibition of PTEN transcription. The PTP domain of PTEN shares similar features with PTP1B phosphatase, presenting the same ability to bind and limit BCR-ABL1 phosphorylation
[66] (Table 1).
 +1 point
+1 point
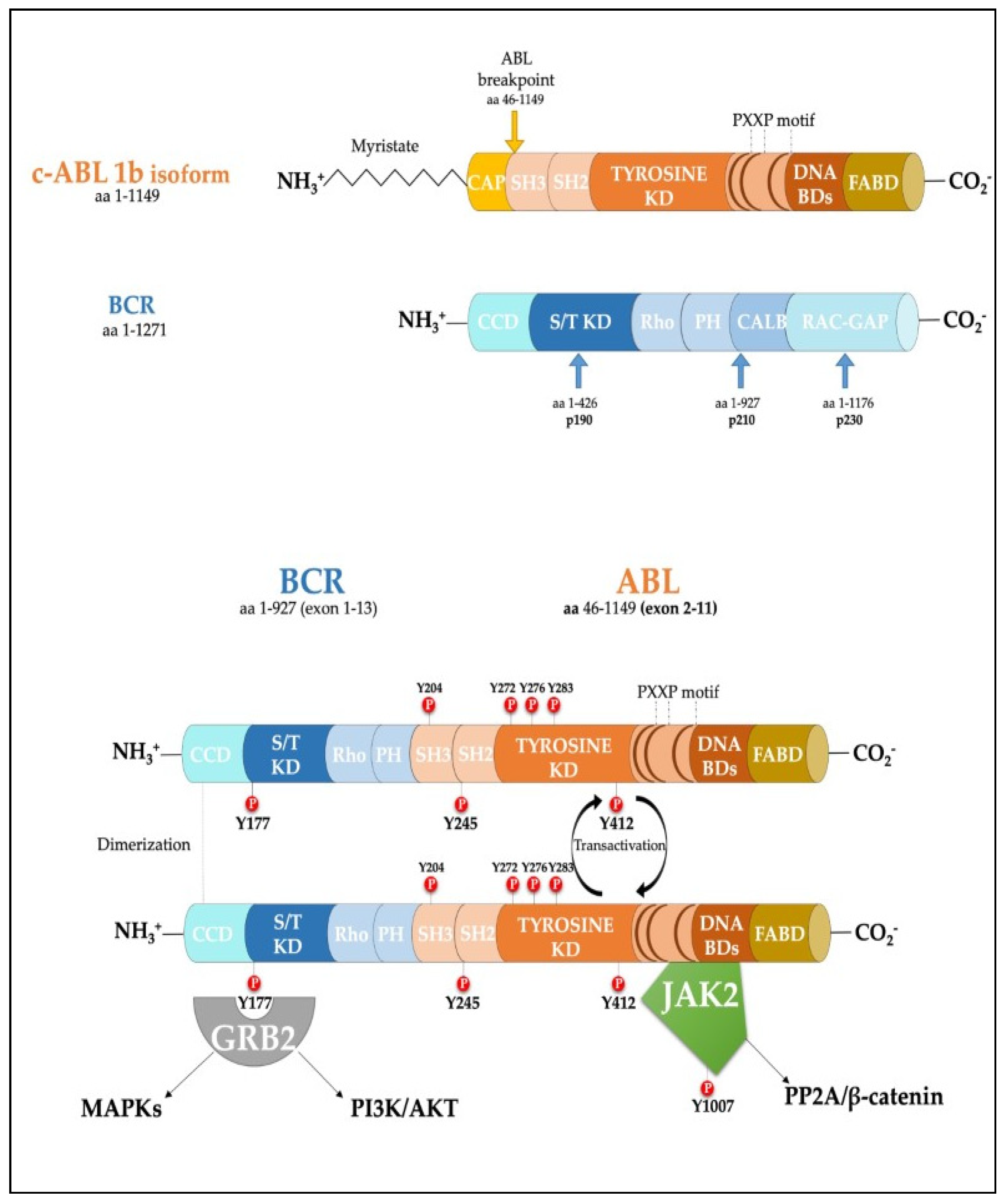 Figure 1. Scheme of the protein products derived from the BCR, ABL1b genes and the fusion product of the BCR-ABL1 oncogene. Major tyrosine phosphorylation sites are represented together with two well characterized interactors relevant for CML disease. SH Src homology domain; PTB Phosphotyrosine-binding domains; KD Kinase Domain; PH Pleckstrin homology domain; BD Binding Domain; FABD f-actin binding domain; CCD Coiled-coil domain.
Figure 1. Scheme of the protein products derived from the BCR, ABL1b genes and the fusion product of the BCR-ABL1 oncogene. Major tyrosine phosphorylation sites are represented together with two well characterized interactors relevant for CML disease. SH Src homology domain; PTB Phosphotyrosine-binding domains; KD Kinase Domain; PH Pleckstrin homology domain; BD Binding Domain; FABD f-actin binding domain; CCD Coiled-coil domain.