Based on the source, ROS production can be divided into exogenous and endogenous production. Exogenous ROS are formed via stimulation by exogenous factors, such as radiation and drugs. Endogenous ROS are produced by mitochondrial and NADPH oxidase (NOX) pathways
[8]. Mitochondria are the main site of ROS production in most eukaryotic cells. During aerobic respiration, most electrons are transported along the respiratory chain and combine with molecular oxygen to form water. However, a small portion of electrons leak out of respiratory chain enzyme complexes I and III, resulting in single-electron reduction in molecular oxygen to form strongly oxidative superoxide anions, which generate hydroxyl radicals and H
2O
2 through specific chemical reactions
[9]. NOX, which is localized on the cell membrane, is a major source of ROS
[10]. External signals, such as bacterial lipopolysaccharide, tumor necrosis factor-α (TNF-α), and interleukin (IL)-1, can stimulate rapid activation of NOX, resulting in a substantial increase in oxygen consumption by cells and a reduction in oxygen molecules to superoxide anions. Superoxide anions are catalyzed by dismutase to generate H
2O
2, leading to a rapid increase in ROS levels to eliminate invading pathogenic microorganisms. In addition, the endoplasmic reticulum (ER) and some enzymes, such as lipoxygenase, cyclooxygenase, and xanthine oxidase, can also generate ROS through chemical reactions
[11].
Organisms have a complete antioxidant system, which is divided into enzymatic and nonenzymatic antioxidant systems. Enzymatic antioxidant systems include superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx). The main function of these antioxidant enzymes is to catalyze the degradation of peroxides. Among them, SOD and CAT are important components of the intracellular antioxidant defense system. SOD includes copper (Cu)/zinc (Zn)-SOD in the cytoplasm and nucleus and manganese (Mn)-SOD in mitochondria. SOD can catalyze superoxide anions to generate H
2O
2 and O
2. The main function of CAT in peroxisomes is to catalyze H
2O
2 to generate O
2 and H
2O
[11]. GPx is an important intracellular enzyme, and in most cases, its activity depends on the micronutrient cofactor selenium. Thus, GPx, which is often referred to as a selenocysteine peroxidase, can decompose H
2O
2 into O
2 and H
2O. GPx has a critical role in inhibiting lipid peroxidation and therefore protects cells from oxidative stress
[12]. Nonenzymatic antioxidants include glutathione (GSH), thioredoxin (Trx), vitamin C, vitamin E, carotenoids, flavonoids, melatonin, and other compounds
[13]. These nonenzymatic antioxidants either scavenge free radicals by acting as hydrogen donors to provide hydrogen ions or alleviate oxidative stress by chelating metal ions, trapping free radicals, and neutralizing peroxyl radicals
[14][15]. Enzymatic and nonenzymatic antioxidant systems act synergistically to provide comprehensive antioxidant protection for cells and tissues.
3. The Role of Redox Dyshomeostasis in the Occurrence of ALL
ALL is a highly heterogeneous hematopoietic malignancy whose etiology and pathogenesis are extremely complex and have yet to be fully elucidated. The occurrence of ALL is currently believed to not be caused by a single factor but may be the result of interactions among various factors, including genetics, infection, ionizing radiation, chemical substances, and immune dysfunction, in a complex environment. Clinical data indicate that most patients with ALL harbor acquired genetic alterations that contribute to the increased proliferation, prolonged survival, or impaired differentiation of lymphoid hematopoietic progenitors. Therefore, ALL can be regarded as a type of genetic disease
[18].
To date, among the more than 200 reported chromosomal abnormalities in leukemias, balanced translocations are the most common, leading to the generation of fusion genes
[19][20]. Abnormal gene expression due to chromosomal translocation or altered function of the encoded fusion protein impairs normal differentiation and yields an aberrant self-renewal capacity, thus having an important role in the malignant transformation of normal hematopoietic stem and progenitor cells (HSPCs). Translocation ETS leukemia-acute myeloid leukemia 1 (
ETV6-RUNX1) is a chimeric transcription factor that is more common in childhood ALL. The incidence of
ETV6-RUNX1 is considerably higher than that of ALL, suggesting the occurrence of additional genetic events during leukemic transformation after birth
[21]. Through the establishment of an
ETV6-RUNX1 transgenic mouse model, Kantner et al. found that although no notable hematopoietic abnormalities were observed in mice, the ROS level in B cells increased, and DNA damage also increased. These results indicated that expression of the oncogene
ETV6-RUNX1 might trigger the second strike by enhancing ROS production, eventually inducing leukemic transformation
[21]. Breakpoint cluster region-Abelson (BCR/ABL) is the most common chromosomal genetic abnormality in adult patients with ALL, with a positivity rate of 20–40%
[22], and encodes proteins with tyrosine kinase activity, promotes cell proliferation, and inhibits apoptosis
[23]. However, the incidence of BCR/ABL-positive samples far exceeds that of the associated leukemias, suggesting that the presence of the BCR/ABL fusion gene alone is insufficient to trigger leukemia
[24]. Several recent studies have found that BCR-ABL oncoprotein-expressing cells are associated with a relative increase in intracellular ROS
[25]. Studies have shown that BCR/ABL can influence ROS production through manipulation of the NOX complex
[26]. In addition, BCR-ABL can also activate the PI3K/AKT/mTOR pathway to promote intracellular ROS production
[25]. Compared with normal cells, BCR/ABL-positive cells suffer more oxidative DNA damage (including DNA double-strand breaks) and demonstrate an increased ability to survive DNA damage. BCR/ABL stimulates the efficiency but decreases the fidelity of the repair mechanisms of double-strand breaks, which may be the leading cause of the accumulation of chromosomal aberrations and subsequent malignant lesions
[27].
To elucidate the role of ROS in the secondary gene events of ALL, Lim et al. recently investigated factors causing mutations in Janus kinase JAK3, JAK1, and Ikzf3 (encoding Aiolos) using a B-cell acute lymphoblastic leukemia (B-ALL) mouse model
[28]. JAKs are nonreceptor tyrosine kinases. Activation of the JAK/signal transducer and activator of transcription (STAT) signaling pathway induces the transcription of genes involved in HSC differentiation and proliferation.
[29]. Aiolos belongs to the Ikaros family and is an essential transcription factor for lymphocyte differentiation
[30]. Abnormal expression of JAKs and Aiolos has been confirmed to be closely related to the development of ALL
[29][31]. Lim et al. found that most mutations with low variant-allele frequency were associated with ROS-induced DNA damage. Application of the JAK inhibitor ruxolitinib can delay leukemia onset, reduce ROS and ROS-induced gene expression signatures, and alter ROS-induced mutational signatures, indicating that JAK mutations can alter the course of leukemia clonal evolution through ROS-induced DNA damage
[28]. The clinical biochemical indices of patients with ALL showed a marked increase in the levels of malondialdehyde, an important biochemical index for plasma oxidative stress
[32], and a notable increase in the levels of 8-oxodG and 8-OHdG, biomarkers of oxidative DNA damage in urine, in patients with ALL
[33][34]. In addition, the level of oxidatively modified DNA bases in lymphocytes from children with ALL was markedly higher than that in children without the disease
[35][36][37]. Abundant evidence has shown that ROS have a vital role in secondary gene events in ALL. However, the specific mechanism underlying the development and progression of ALL remains to be further elucidated.
According to the second hit theory, the occurrence of leukemia is the result of the accumulation of multiple gene abnormalities. Abnormal gene expression activates specific signaling pathways to promote the malignant transformation of cells. PI3K/AKT/mTOR, MAPK kinase (MEK)/extracellular signal-regulated kinase (ERK), and JAK/STAT are the major signaling pathways of oxidative stress and are also the representative signaling pathways for abnormal activation of leukemia cells
[38][39]. In normal HSCs, overactivation of the above oxidative stress pathways promotes the production and intracellular accumulation of ROS, severely disturbs the normal biological functions of HSCs, and has an important role in leukemia progression
[38][39]. Recent studies have shown that abnormal expression of a variety of genes activates the related oxidative stress pathway, thereby affecting ALL progression. These genes mainly include Notch, PTEN, RAS, and IL-7 and its receptor (IL-7R). (1) Notch encodes a transmembrane receptor that regulates normal T cell development. Mutations in this gene are common in T cell acute lymphoblastic leukemia (T-ALL). In T-ALL cells, the withdrawal of Notch signals prevents stimulation of the mTOR pathway by mitogenic factors, indicating that Notch has a positive regulatory role in the mTOR pathway
[40]. Mutant Notch can activate the mTOR pathway through the PI3K/AKT pathway or through activation of c-Myc without relying on the PI3K pathway
[40][41]. (2) PTEN is a tumor suppressor gene closely related to tumor development. Its encoded protein has dual protein/lipid phosphatase activity and is a major phosphatase with a negative regulatory effect on the PI3K/AKT pathway
[42]. Mutation or deactivation of PTEN after translation can result in chronic activation of PI3K/AKT/mTOR signaling in ALL cells, γ-secretase inhibitor (GSI) resistance, and inhibition of p53-mediated apoptosis
[41]. (3) RAS is a proto-oncogene, and its encoded proteins are small GTPases, which act as molecular switches. Activated RAS promotes ROS production through NOX stimulation
[43] and transduces signals from a variety of cell surface receptors to downstream signaling pathways, such as PI3K/AKT/mTOR and MAPK, to regulate a number of cell fate decisions. RAS-activating mutations and Notch mutations synergistically promote the development and progression of T-ALL
[44]. (4) IL-7 and IL-7R are required for normal lymphocyte development. Without them, severe combined immunodeficiency occurs. However, excessive activation of IL-7/IL-7R signaling activates the three oxidative stress signaling pathways, PI3K/AKT/mTOR, MEK/ERK, and JAK/STAT, to promote the development of ALL and the resistance of ALL cells to chemotherapeutic drugs
[28][45]. Lim et al. found that ROS induced by IL-7 in the downstream JAK/STAT pathway positively increased JAK/STAT signaling and that the occurrence of B-ALL and the survival of B-ALL cells were dependent on high levels of ROS driven by IL-7-dependent JAK/STAT signaling
[28]. Silva et al. reported a positive feedback interaction between the IL-7-mediated PI3K/AKT signaling pathway and ROS. Moreover, activation of this pathway upregulated glucose transport protein type 1 and increased glucose uptake by T-ALL cells. The use of ROS scavengers inhibited the viability of T-ALL cells or even led to cell death
[46]. These results suggest that ROS not only drive the development of ALL but are also indispensable for the survival of ALL cells. The major redox signaling pathways associated with ALL transformation are shown in , and the corresponding targeted drugs are shown in .
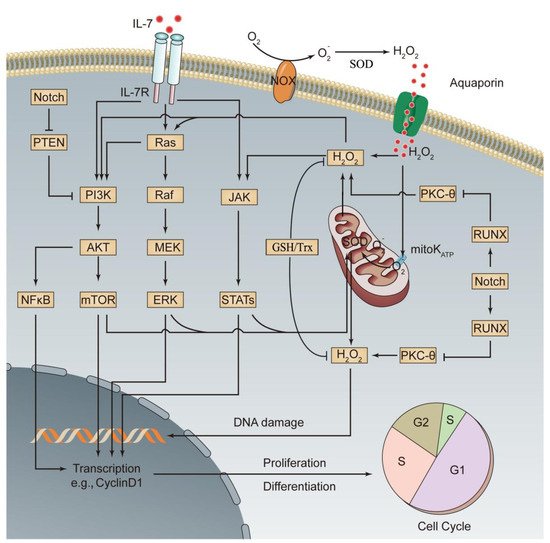
Figure 1. Activated oxidative stress signaling pathways involved in the pathogenesis of ALL. Increases in oncogene-, chemical drug-, or radiation-induced ROS production or abnormal expression of relevant genes leads to activation of three major oxidative stress signaling pathways, PI3K, MEK, and JAK, thus promoting the differentiation and proliferation of leukemia cells. Additionally, activation of oxidative stress signaling pathways promotes mitochondrial ROS production through enhanced oxidative metabolism, which further activates the three oxidative stress signaling pathways, thereby forming a positive feedback signaling pathway. A series of mechanisms in leukemia cells prevent excessive ROS production, thus avoiding cell injury or death (see the text for details). This figure was drawn based on existing research data; its accuracy and more precise signaling mechanisms must be confirmed and supplemented by extensive, in-depth studies. mitoKATP, mitochondrial ATP-sensitive K+ channel.
Table 1. ALL redox signaling pathway targets and representative drugs.
Signaling
Pathway |
Targets |
Representative Drugs |
Antileukemic Effect |
Refs |
| BCR/ABL |
Tyrosine kinase inhibitor (TKI) |
Imatinib |
First-generation TKI that can block the ATP-binding sites of BCR-ABL and prevent activation of the conformation of oncogenic proteins |
[47] |
| Nilotinib |
Second-generation TKI and high-affinity aminopyrimidine-based ATP-competitive inhibitor with more specific inhibition of BCR/ABL activity |
[48] |
| Dasatinib |
Second-generation TKI that can bind to inactive and active BCR/ABL kinase and inhibit Src family kinases and c-Kit |
[48] |
| Bosutinib |
Third-generation TKI and potent dual inhibitor of Src and ABL kinases with longer-term safety than second-generation and other third-generation TKIs |
[49] |
| Ponatinib |
Third-generation TKI that is effective for known mutations in imatinib-resistant genes (including T315I) |
[50] |
| Notch |
γ-secretase inhibitor (GSIs) |
BMS-906024 |
Inhibits the activity of Notch signaling by downregulating the expression of multiple known target genes of Notch but has no marked effect on c-Myc |
[51] |
| PF-03084014 |
Downregulates the level of the Notch intracellular domain and the expression of Notch target genes Hes-1 and c-Myc and induces cell cycle arrest and apoptosis of T-ALL cells |
[52] |
| PI3K/AKT/mTOR |
PI3K-δ inhibitor |
Idelalisib |
Downregulates the level of AKT phosphorylation in B-ALL cells, inhibits cell proliferation, and blocks the homing of B-ALL cells into the bone marrow |
[53] |
| NVP-BKM120 |
Downregulates the phosphorylation levels of AKT and mTOR in T-ALL cells, inhibits cell cycle progression, and promotes apoptosis |
[54] |
| AKT inhibitor |
MK-2206 |
Downregulates AKT phosphorylation levels in both T-ALL and B-ALL cell lines (it can also promote PTEN phosphorylation in B-ALL cell lines), inhibits cell proliferation, and promotes apoptosis |
[55] |
| PI3K/mTOR inhibitor |
PI-103 |
More potent than inhibitors that are selective only for PI3K or for mTOR and can effectively induce cell cycle arrest and apoptosis in T-ALL cells |
[56] |
| JAK/STAT |
JAK inhibitor |
Ruxolitinib |
JAK1/2 inhibitor that can reduce ROS and ROS-induced gene expression signatures and inhibit the growth of leukemia cells |
[28] |
| RAS |
MEK inhibitor |
Selumetinib
Trametinib
MEK162 |
Reduce ERK phosphorylation and induce apoptosis in the RAS-mutant MLL-rearranged ALL cells |
[57] |
4. Redox Regulation in Leukemia
The hypoxic BM niche is not only a habitat for HSCs but also a natural sanctuary for malignant blood cells. Leukemia cells hidden in the BM niche can avoid the cytotoxicity of chemotherapeutic drugs, resulting in the formation of minimal residual disease. Minimal residual disease is an accepted root cause of drug resistance and relapse in leukemia
[58]. A large body of evidence has shown that in a hypoxic environment, the ROS level and its regulation have important roles in leukemia cell survival, proliferation, differentiation, immune escape, and epigenetic changes
[59].
Similar to HSCs, primitive leukemia cells also have self-renewal, self-differentiation, and self-proliferation capacities, and their relatively low intracellular ROS levels are conducive to cell survival and stemness maintenance
[60][61]. Compared with that in leukemia cells with higher proliferating activity, the ROS level in primitive leukemia cells is lower. This phenomenon has been observed in many types of leukemia cells
[2][61]. Giambra et al. also observed that the most aggressive leukemia-initiating cells (LICs) in T-ALL model mice and human T-ALL were characterized by low levels of ROS, and that the increase in ROS levels inhibited the activity of LICs
[62], suggesting that the behavior of ALL cells is closely related to ROS levels and oxidative stress status.
Similar to other types of cancer cells, leukemia cells can also undergo glycolysis for energy metabolism. However, a large amount of evidence indicates that leukemia cells mainly gain energy through mitochondrial respiration and that ROS produced through the mitochondrial respiratory chain thus become a major source of ROS in leukemia cells
[63][26]. Markedly improved NOX activity has been observed in many leukemia cell lines, including AML, chronic myelogenous leukemia, and promyelocytic leukemia cell lines, indicating that constitutive activation of NOX is another important source of ROS in leukemia cells
[64][65][66]. In the in vitro culture of T-ALL cells, the inhibition of complex I of the respiratory chain with rotenone and the disassembly of NADPH subunits with apocynin both abrogated the IL-7-mediated elevation of intracellular ROS levels, confirming that the mitochondrial respiratory chain and NOX are also the main sources of intracellular ROS production in T-ALL cells
[46]. Studies have found that in lymphoblastic leukemia cells, xanthine dehydrogenase and xanthine oxidase can oxidize NADH to catalyze the production of ROS, suggesting that in addition to the mitochondrial respiratory chain and NOX complex, certain metabolic/detoxification pathways in lymphoblastic leukemia cells are also important sources of ROS production
[67]. In addition, as described above, overactivation of three signaling oxidative stress pathways, PI3K/AKT/mTOR, MEK/ERK, and JAK/STAT, and oncogenes such as BCR/ABL and RAS are related to the changes in redox homeostasis in leukemia cells and increases in ROS levels.
Previous studies have shown that decreased antioxidant defense exists in multiple types of leukemia
[68][69][70]. Therefore, redox dysregulation may be one of the causes of the increase in ROS levels in leukemia cells. Sentürker et al. conducted a controlled study with a group of untreated children with newly diagnosed ALL and normal children and confirmed that three antioxidant enzymes, CAT, GPx, and SOD, in blood lymphocytes in children with ALL were lower than those in normal children in the control group
[36]. Battisti et al. evaluated the oxidative status and antioxidant defense of patients with ALL and found that CAT and SOD activities in the whole blood of patients with ALL were lower by different levels than those in the normal control group and that SOD activity was lowest in newly diagnosed patients
[68]. However, Ben Mahmoud et al. found that the plasmatic activities of CAT and SOD and the plasma levels of reduced GSH were elevated in patients with ALL and that SOD activity and GSH levels were substantially correlated with ALL relapse
[71]. Nishiura et al. found that serum levels of Mn-SOD in patients with ALL were substantially higher than those in normal controls, but as the disease subsided, serum levels of Mn-SOD decreased
[72]. Although the specific causes leading to large differences in the results between different studies are still unclear, the existing research findings suggest that changes in the antioxidant defense capacity of ALL cells may be related to the development and progression of the disease and the course of treatment. At the onset of leukemia, the decreased antioxidant defense may be related to the occurrence of events such as oxidative DNA damage and genomic instability. During the progression and treatment stages of leukemia, increased antioxidant defense may be an adaptive response to the increase in oxidative stress, enabling leukemia cells to survive under high levels of oxidative stress and to even antagonize the cytotoxic effects of chemotherapy drugs and develop drug resistance.
In the development and progression of leukemia, on the one hand, malignant transformation of HSCs must be initiated through the production of high levels of ROS; on the other hand, continuous intracellular ROS accumulation must be prevented to avoid oxidative damage or cell death, which requires real-time and dynamic regulation of intracellular oxidation and antioxidant status. This regulation process not only depends on the participation of the intracellular antioxidant defense system but also involves a series of other complex intracellular and extracellular mechanisms. The GSH redox system represents one of the most important cellular defense systems against oxidative stress, and high intracellular GSH levels have been linked to treatment resistance in leukemia cells
[73]. The Trx system is another major cellular antioxidant network and is composed of NADPH, Trx reductase, and Trx, which can provide electrons for thiol-dependent peroxidases (peroxiredoxins) to remove reactive oxygen and nitrogen species with a fast reaction rate, thereby protecting cells from oxidative damage
[1][15]. For relatively quiescent LICs, regulation of the intracellular oxidative stress status is more important for the maintenance of their invasiveness. As mentioned above, Notch is frequently activated by mutations in T-ALL, and activated Notch participates in leukemic transformation by activating the mTOR oxidative stress pathway. However, Notch also downregulates the expression of protein kinase C θ (PKC-θ) through the runt-related transcription factor (RUNX) pathway, thereby inhibiting intracellular ROS accumulation and facilitating the maintenance of LIC activity
[62]. In addition, numerous studies have shown that various antioxidant proteins, such as nuclear factor erythroid 2-related factor 2 (Nrf2) and heme oxygenase-1 (HO-1), also have important regulatory roles in the antioxidant responses of ALL cells
[74][75].
Extracellularly, ALL cells establish extensive contact with the BM niche via chemokines, adhesion molecules, and exosomes and transform the BM niche into a leukemia growth-permissive and normal hematopoiesis-suppressive leukemia niche to improve the survival of ALL cells in a hypoxic environment
[76]. Under oxidative stress conditions, BMSCs produce protective soluble factors, which help ALL cells achieve redox adaptation and reduce oxidative stress damage, ultimately inducing the reversible multidrug resistance phenotype in ALL cells
[5]. Tunneling nanotubes (TNTs), which were discovered by Rustom et al. in 2004 using a three-dimensional live cell microscopy, serve as an intercellular communication method
[77]. Recent studies have found that ALL cells regulate the BM niche through the use of TNTs to induce the secretion of prosurvival cytokines by signaling to primary MSCs, thereby contributing to the survival of ALL cells
[4]. Mitochondria in ALL cells have also been found to be transferred to MSCs through TNTs to alleviate oxidative stress, thereby reducing intracellular ROS levels and avoiding chemotherapeutic drug-induced death
[3]. Therefore, the BM niche inhabited by ALL cells has become an important sanctuary for maintaining redox homeostasis and resisting oxidative stress damage. Breaking the antioxidation barrier of leukemia cells is currently an important topic in the promotion of pro-oxidant therapy for leukemia and has received extensive attention from researchers.
 +1 point
+1 point