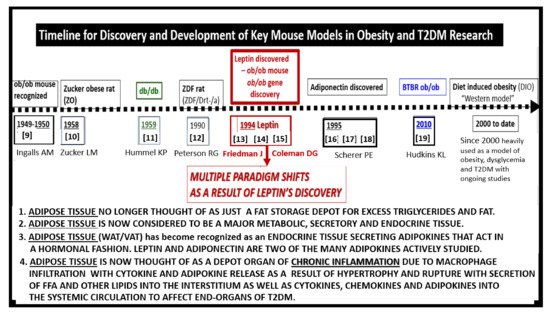
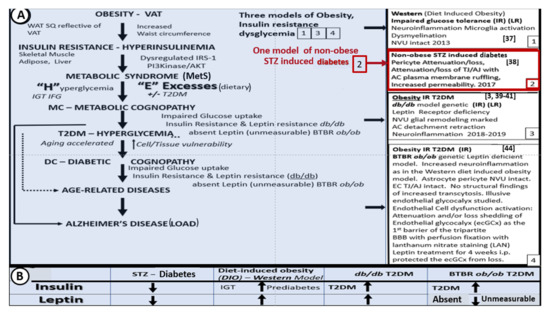
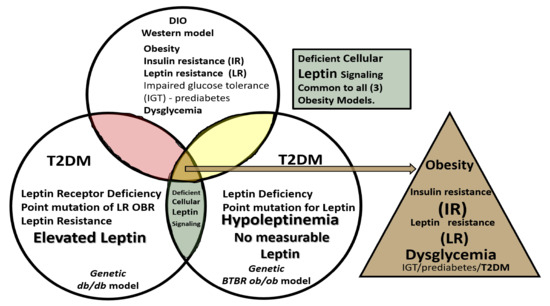
5. Microglia (MGC)
As noted above, aMGC encroaching into the NVU and encompassing the BEC are a frequent finding in the BTBR
ob/ob model in the hippocampus. In the cortex, ramified microglia were increased compared to controls, but there was no activation in cortical Layer III of microglia. Ramified microglial cells (rMGCs) protect brain functions by phagocytosing cellular debris. MGCs are of mesodermal (yolk sac derived) origin and serve the brain as resident innate immune cells. They are responsive to many cytokines, chemokines, and signaling molecules, producing free radicals (superoxide, reduced nicotinamide adenine dinucleotide phosphate (NADPH Ox), inducible nitric oxide and mitochondrial-derived reactive oxygen/nitrogen (mtROS). The aMGCs are able to return to their surveilling-ramified phenotype once the invaders or danger–damage signals have been eradicated. Additionally, MGCs are important in brain development and play an important role shaping and pruning neuronal synaptic connectivity in formative and adult years
[41][45][46][47][48][49][50][51][52][53][54][55]. MGCs are distinct from bone marrow derived peripheral monocyte-macrophage cells in that they are the brain resident inflammatory immune cells and not dependent on recruitment from the peripheral systemic circulation but are capable of undergoing proliferation and activation as needed
[56]. Multiple toxicities in obesity and T2DM including the absence of leptin cellular signaling may result in aMGCs in the
db/db models to a much greater extent as compared to the BTBR
ob/ob models ()
[3][41][43].
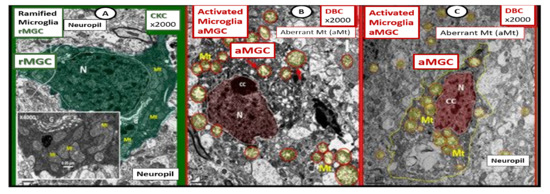
Figure 5. Comparison of ramified microglia (rMGC) in control mice to activated microglia (aMGC) in obese Type 2 diabetes mellitus (T2DM)
db/db model.
Panel A illustrates the normal appearance of ramified microglia (rMGC-pseudo-colored green) (see A,C). In addition, note the insert in
Panel A and the presence of cristae in rMGC at higher magnification ×6000).
Panels B and
C depict aMGCs (pseudo-colored red) with swollen electron lucent aberrant mitochondria (aMt) (pseudo-colored yellow) the
db/db diabetic mice. Magnification ×2000; scale bar = 1 µm in
A and
B and 2 µm in
Panel C. These images are adapted with permission from references
[3][39]. CC = chromatin condensation; CKC = control C57BL/6J female non-diabetic model; DBC = female diabetic
db/db model.
We observed an increase in the number of rMGCs in the cortical Layer III of the BTBR ob/ob as compared to control models and this may be due to an increase in metabolic neuronal and NVU activity in these regions. In the hippocampal regions of the BTBR ob/ob, we not only observed a marked increase in rMGCs in excess of the cortical regions as compared to control models, but also noted the encroaching aMGCs ().
Our results in the BTBR
ob/ob model strongly suggest that these models are a pro-neuroinflammatory phenotype, especially in the hippocampal regions. Interestingly, the difference between the level of activation of the microglia in the
db/db and BTBR
ob/ob suggests a difference in their genetic background even though both models were obese and developed T2DM with hyperglycemia at 20-weeks. In the original publication describing the BTBR
ob/ob model, Hudkins et al.
[19] noted that glucose levels were in the 300 mg/dL range and that in the
db/db the mean glucose at a similar age were in the range of 500 mg/dL
[3][41][43][57][58]. This difference in the amount of glucotoxicity could be at least one reason the activation of MGC were not as prevalent in the female BTBR
ob/ob as compared to the
db/db models
[19]. Additionally, BTBR
ob/ob fetuses, unlike
db/db fetuses, will have leptin stimulation probably until weaning. Ultrastructural observations in the
db/db and BTBR
ob/ob certainly implicate the activation of resident microglia and encroachment of the NVU by aMGCs may be one of the mechanisms for impaired NVU capillary dilation due to regional neuron signal or NVU uncoupling associated with decreased regional cerebral blood flow within the hypothalamus
[3][41]. This NVU uncoupling could certainly be related to a decrease in CBF and neuronal ischemia resulting in synaptic dysfunction and/or neuronal loss with ensuing impaired cognition and regional brain atrophy. However, brain atrophy has only been reported in the
db/db models by Ramos-Rodrigues et al.
[59] and was not found to be present in this current BTBR
ob/ob study ().
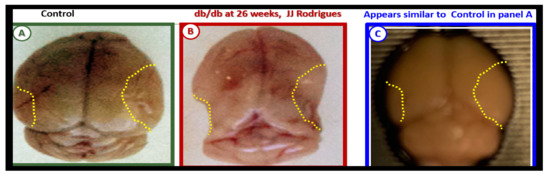
Figure 6. Gross brain atrophy in the
db/db model but not in the diabetic BTBR
ob/ob model.
Panel B illustrates the brain atrophy at the time of surgical removal of the diabetic
db/db models as compared to control models in
Panel A.
Panel B depicts the marked atrophy or loss in the cortical-parietal-hippocampal regions (outlined by the yellow dashed lines) in comparison to the control in
Panel A at 26-weeks.
Panel C demonstrates the BTBR
ob/ob model at 20-weeks without atrophy in the parietal-hippocampal regions of the whole brain when compared to the
db/db model in
Panel B at 26-weeks. This may correlate with the less severe ultrastructural remodeling of the neurovascular unit and less microglial neuroinflammation in BTBR
ob/ob mice as compared to
db/db mice.
Panels A and
B CC by NC-ND
[59].
Ramos-Rodriguez et al.
[59] have demonstrated that the
db/db models have abnormal gross morphological findings with decreased brain weights, including grossly evident cortical parietal-hippocampal atrophy, a finding not present in the BTBR
ob/ob model (). These findings suggest an interaction of genetic background with obesity and T2DM in regards to brain morphology as well as the role of leptin receptor deficiency vs deficient leptin production as in the BTBR
ob/ob models. Whether this atrophy relates to differences in neurogenesis, decreased CBF, or neuronal ischemia with loss of synapses and neurons is unknown.
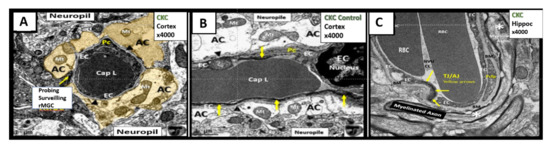
6. Blood-Cerebrospinal Fluid Barrier (BCSFB), Choroid Plexus, Median Eminence of Third Ventricle, Tanycytes, Circumventricular organ(s) (CVOs) and Hypothalamic Nuclei
The brain’s ventricular system (two lateral and third, aqueduct, and fourth ventricles) contain the cerebrospinal fluid (CSF) and are lined primarily by cuboidal epithelial cells termed ependymal (EPY) cells ()
[60].
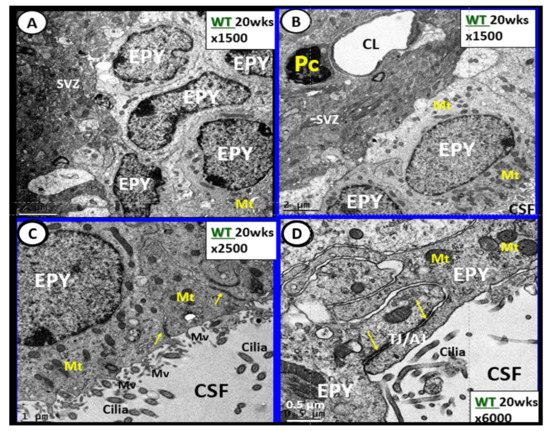
Figure 7. Ependymal cells lining the ventricular cerebrospinal fluid (CSF) system of the aqueduct in female control 20 week old models. Panel A illustrates that not all ependymal cells (EPY) are cuboidal but may lie flattened and be multilayered at the subventricular zone (SVZ) region of the adjacent neuropil. Panel B demonstrates the more classical cuboidal morphologic phenotype of the EPY cells that line the CSF ventricles. Note the close 5 µm distance between the EPY cells and fenestrated capillary within the SVZ neuropil. The adjacent capillary neurovascular units (NVU) in the SVZ do not have a similar coverage by astrocytes as in the cortical regions but pericytes (Pc) are frequently noted and this capillary appears to be fenestrated due the thinning of the ECs. Panel C depicts the ependymal cilia with the classical 9:2 arrangements of cytoskeletal proteins within them in addition to multiple microvilli (Mv) on the apical surface facing the CSF. Panel D demonstrates the overlapping and interdigitations of EPY cells, and also the staining of the very electron dense tight and adherens junction (TJ/AJ) proteins (arrows) and desmosomes that create the barrier functions of the EPY cells in some regions of the ependymal lining of the CSF. While these EPY cells are located in the aqueduct they also are representative of the EPY cells found throughout each of the four ventricles and lining of choroid plexus (CP) that provides the CP with its barrier function. As noted in these images, the EPY cells form the structural and functional barrier of the choroid plexus. CL = capillary lumen.
EPYs have CSF apical microvilli that increase their surface area and cilia to aid the movement of CSF within the ventricles. EPYs have a genetic linkage to the neuroepithelial cells of the developing neural tube of the embryo. CSF within the ventricles is thought to serve as a sink for the deposition of toxic metabolic byproducts created by the high metabolic neuronal activity within the brain and their subsequent removal into the systemic venous pathway (including the toxic amyloid beta proteins). The CSF from the ventricles enters the subarachnoid space to be drained by the subarachnoid granulations and nasal lymphatics into the systemic circulation
[61][62][63][64][65].
The rather uniform coverage of the ventricles by EPYs gives way to a specialized covering of the choroid plexus (CP) in the floor of the lateral ventricles and the roof of the third ventricle. The CP is supplied by the choroid arterial system and these end in a plexus of capillaries that are fenestrated. These fenestrated capillaries produce a plasma filtrate that is separated from the CSF by the EPYs that form the CP. Importantly, these EPYs of the CP possess TJ/AJ and secrete the bulk of CSF ()
[66][67].
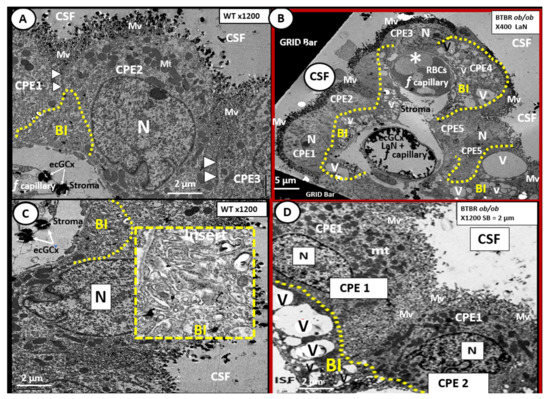
Figure 8. Choroid plexus (lateral ventricle) with aberrant vacuolization of the basilar infoldings of ependymal cells in obese diabetic female BTBR ob/ob models. Panel A demonstrates the normal ultrastructural morphology of the choroid plexus ependymal cell(s) (CPE) in the control heterozygous model, characterized by compact basilar infoldings (BI) (yellow dashed line), tight and adherens junctions (TJ/AJ) (arrowheads), microvilli (Mv) of the brush border at the apical cerebrospinal fluid (CSF) interface, and the multiple electron dense mitochondria (Mt) in a highly polarized ependymal cell. Panel B depicts aberrant remodeling changes of the (BI) at lower magnification to include multiple CPEs (CPE 1–5) which consist of vesiculation (v)/vacuolization (V) of the BI (yellow dashed lines). Importantly, note the lower fenestrated (f) capillary with positive staining for lanthanum nitrate (LaN) of the endothelial glycocalyx (ecCGx) and the upper capillary (*) that is filled with multiple red blood cells (RBCs) that is highly suggestive of capillary micro-thrombosis and without evidence of LaN staining. Panel C also illustrates the control heterozygote CPEs and this image contains an exploded insert of the BI (outlined in yellow dashed lines). This insert illustrates the compactness of the BIs without v/V. Panel D depicts the v/V of the BI as compared to the control models in Panels A and C. Magnification ×400; scale bar = 5 µm in Panel B and magnification ×1200; scale bar = 2 µm in Panels A–D. n = nucleus.
The CP is primarily responsible for two-thirds of the CSF production along with the EPY cells and cells lining the subarachnoid space
[68]. Of significant importance is the recent report that insulin is synthesized in the EPY cells of the choroid plexus, which is regulated by serotonergic signaling
[69]. The CP regions are covered by the EPY cells as previously imaged and each of these adjacent EPY cells are known to contain TJ/AJ, forming the barrier function of the CP and the BCSFB ( and ).
As one traces the ciliated EPY cells to the median eminence of the hypothalamus, the EPY cells give way to tanycytes, which are highly specialized ependymal cells, and lose their cilia, supposedly to better sense the multiple hormonal and non-hormonal proteins and polypeptide contents in the CSF. Importantly, the tanycytes also have elongated subventricular zone (SVZ) cytoplasmic processes that may directly interact with the portal fenestrated capillaries of the CVOs and are also capable of connecting the median with the hypothalamus.
Tanycytes were originally documented and named by Horstmann in 1954
[70]. Further, tanycytes have been discussed in detail by Rodriguez et al.
[71] with more recent discussions provided by Gao et al. and Balland et al.
[72][73], as well as in regards to the neuroimmune axis of the barriers
[73][74]. Currently there are thought to be at least four types of tanycytes (alpha1, 2 and beta 1, 2-dorsal β1d, ventral β1v, lateral β2la and medial β2me) which have varied functions. The tanycytes are not only responsible for glucose sensing via their glucose transporters but also leptin sensing via its leptin receptors (LepRs)
[74][75]. As one follows the lining EPYs of the CSF ventricles to the region of the median eminence the lining EPYs differentiate to become tanycytes, which are one mechanism for the uptake and distribution of leptin to the hypothalamus.
Intraventricular CSF lining tanycytes are highly specialized bipolar ependymal cells that line the ventrolateral wall and the floor of the third ventricle and play a significant role in leptin uptake at the level of the median eminence of the hypothalamus. Tanycytes also play a key role in our obesity and T2DM models in this review (DIO Western,
db/db and BTBR
ob/ob) as they sense leptin and glucose
[76][77]. Therefore, tanycytes play a necessary and essential role in regulating peripheral metabolic signals and energy balance since leptin is considered to be the afferent arm of the brain’s negative feedback control of this delicate balance between peripheral WAT adiposity and the CNS response
[75][76]. The above dual vascular supply of leptin via the pial BBB capillaries and the choroid capillaries of the CP; BCSFB are very important for leptin brain hypothalamic signaling and maintaining leptin CNS homeostasis
[66][78].
In the BTBR ob/ob females, we found evidence of remodeling of the ventricular system (). In particular, the basilar infoldings were highly vacuolated and vesiculated. The presence of microthrombosis illustrates the risk of vascular to the CP as well as to brain parenchyma.
 +1 point
+1 point