1000/1000
Hot
Most Recent
 +1 point
+1 point
Apoptosis is a multistep process that involves two major pathways to trigger a cascade of events leading to the fragmentation of chromatin, nuclear membrane, and cell shrinkage. However, when this physiological process tended to be dysregulated, many pathological transformations happen to develop cancer.
The term “apoptosis” is derived from a Greek word with the meaning of falling off of the dead leaves from trees in autumn [1]. Kerr et al. first proposed this term to describe morphological alterations of the cells during this process. These morphological changes occur through two main steps. The early apoptosis events include pyknosis (i.e., decreased cell volume), nuclear degradation, and chromatin condensation [2]. At the later step of apoptosis, initial morphological changes in the cells include cell shrinkage, plasma membrane blebbing, cytoplasmic organelles modification, loss of cell membrane integrity, and production of apoptotic bodies [3]. The process of apoptosis was identified very late in the history of cellular biology; because the apoptotic cells are usually engulfed by phagocytes before the formation of apoptotic bodies. The presence of apoptotic bodies was discovered in vitro under particular conditions. Under these conditions, the remnants of apoptotic cells generally undergo degradation, which is known as secondary necrosis [4].
Early in apoptosis, phosphatidylserine (PS) molecules usually are flipped out to the outer layer of the cell membrane. PS exposure to the cell surface displays an engulfment signal, which attracts phagocytic cells to engulf apoptotic cells without the secretion of inflammatory cytokines [5]. It has been clearly described that various endogenous and exogenous agents trigger programmed cell death in a specific cell type. Physical stimulators (such as radiation, trauma, and chemotherapeutics) and infectious pathogens (viruses and bacterial toxins) are exogenous factors that affect most types of cells. Endogenous activators of apoptosis include the absence of growth factors, trophic hormone deficiency, glucocorticoid therapy, and ablation of matrix attachment [6]. Regardless of the type of stimulators of apoptotic cell death, this process is usually triggered through two distinct mechanisms and pathways that are completely explained in the next section. Figure 1 depicts major apoptotic pathways in relevant factors.
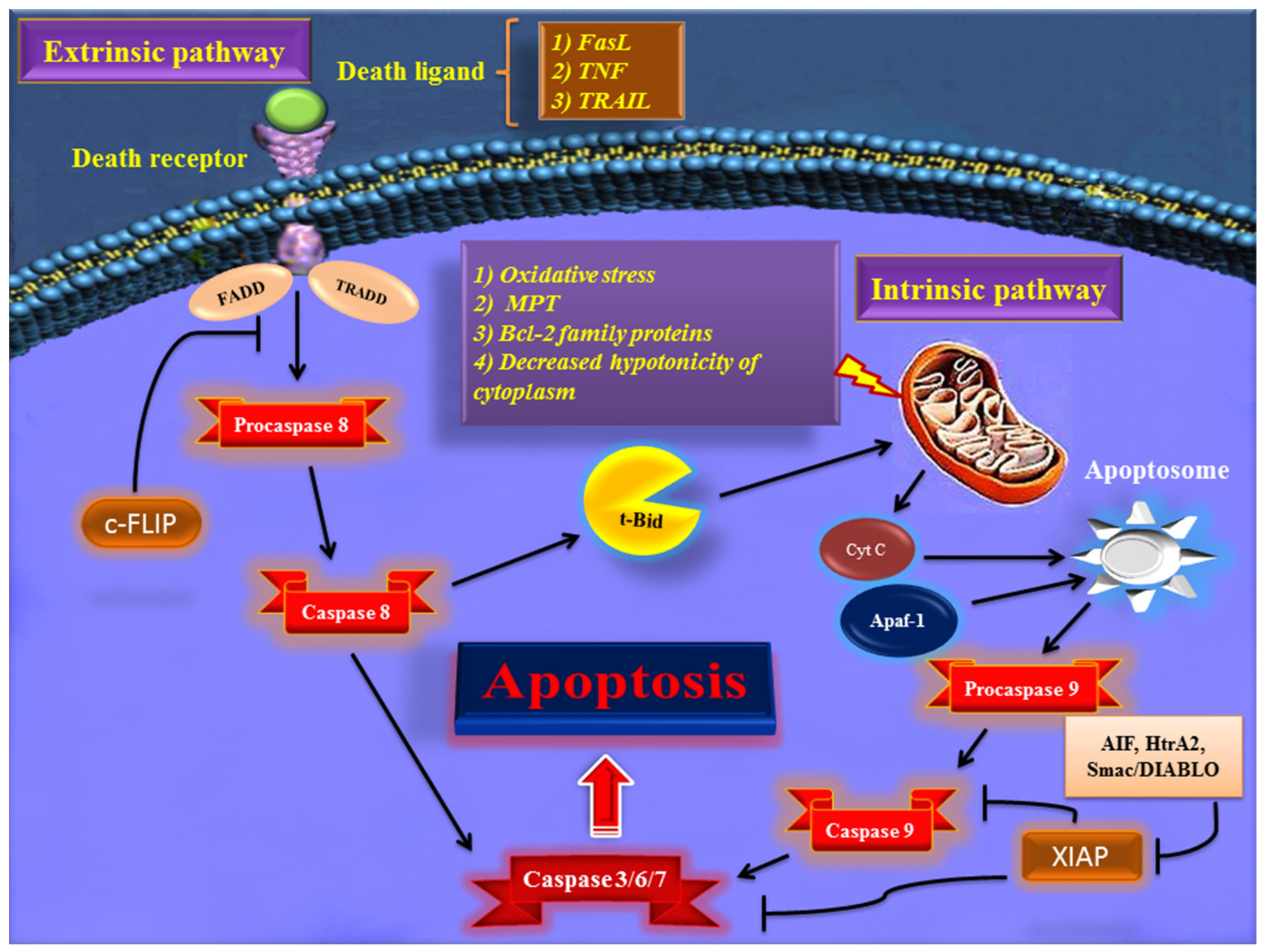
Figure 1. Overview of the extrinsic and intrinsic apoptosis signaling pathways and their major role-players. FasL—Fas ligand; TNF—tumor necrosis factor; TRAIL—TNF- related apoptosis-inducing ligand; TRADD—TNF receptor-associated death domain; FADD—Fas-associated death domain; Bcl-2—B cell lymphoma-2; Cyt c—cytochrome c; Apaf-1—apoptotic protease activating factor 1; AIF—apoptosis-inducing factor; Smac/DIABLO—second mitochondria-derived activator of caspase/direct IAP binding protein with low pI; HtrA2—Omi/high-temperature requirement protein A; XIAP—X-linked inhibitor of apoptosis protein; c-FLIP—cellular FADD-like IL-1β-converting enzyme (FLICE) inhibitory protein; MPT—mitochondrial permeability transition; t-BID—truncated BH3 interacting-domain death agonist.
One of the main hallmarks of cancer is cell death evasion [7]. Several stressor stimuli induced by cancerous conditions compel cancer cells to avoid apoptosis, leading to the development of a tumor and resistance to therapy [8]. Cancer cells elaborate several mechanisms to dysregulate intrinsic or extrinsic pathways to evade apoptosis (Figure 2).
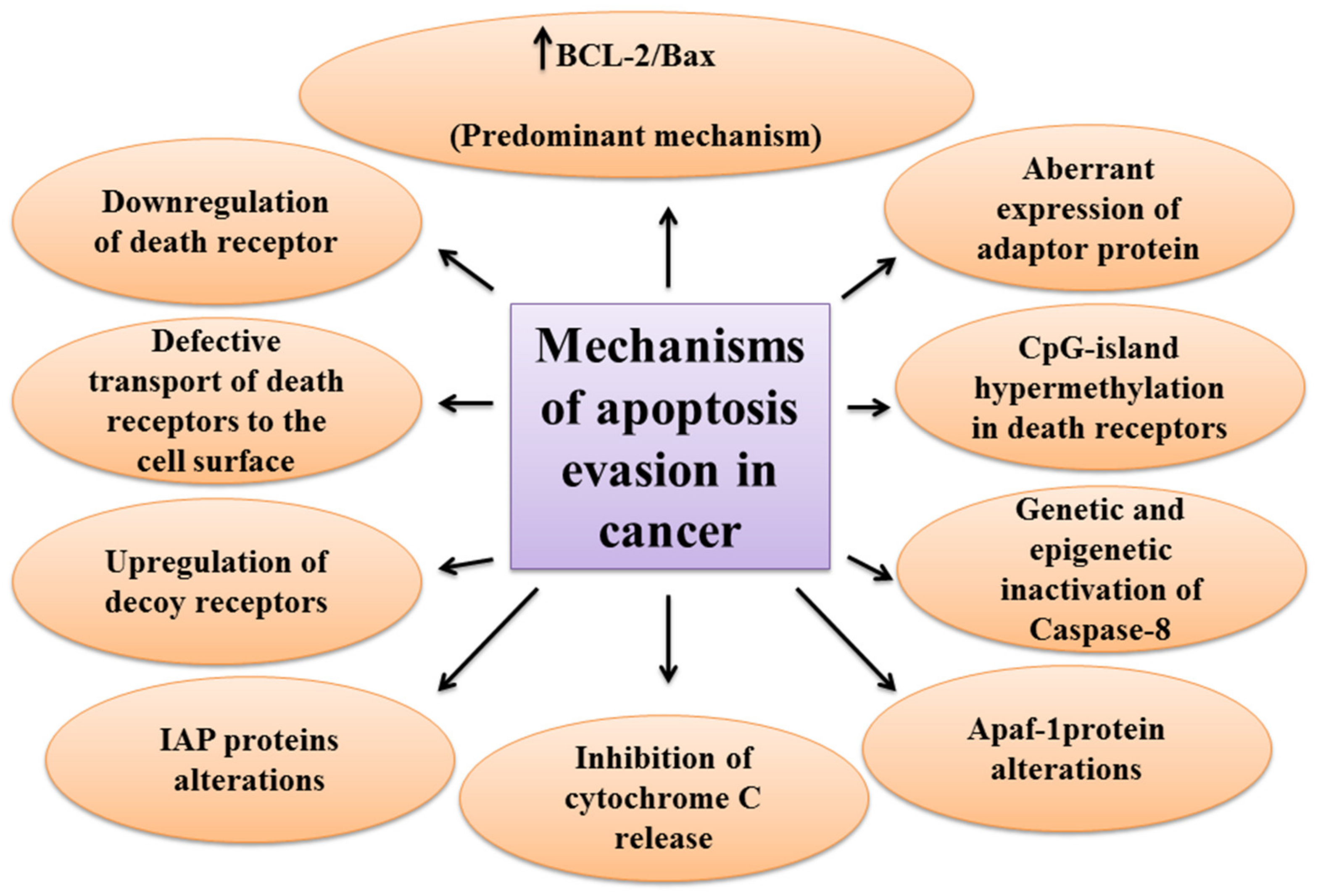
The first molecules that are impaired in the extrinsic pathway are death receptors. For instance, downregulated CD95, as a death receptor, has been attributed to the resistance of leukemia and neuroblastoma cell lines to treatment approaches [9]. Furthermore, defective molecules that act downstream of the death receptors may contribute to the development of apoptosis resistance in tumors.
Tourneur et al. found that downregulation or loss of expression of the FADD protein in leukemic cells was a predictor of resistance to chemotherapy and a poor prognostic factor [10]. Treatment modalities targeting TRAIL receptors revealed that the main cause of apoptosis resistance in colon cancer cell lines is the inappropriate transport of DR-4 and DR-5 receptors from intracellular sources to the cell surface [11]. Further, epigenetic mechanisms like CpG-island hypermethylation can hamper the expression of death receptors and lead to the decreased levels of the receptors in the cell membrane [12]. The DcRs are another way for cancers to evade apoptosis. For example, DcR3, which binds Fas ligand by a competitive mechanism and blocks Fas ligand-mediated apoptosis, was identified to be upregulated in glioblastoma and carcinomas of lung and colon [13]. In the meantime, upregulation of c-FLIP, an antiapoptotic factor, has been shown in numerous cancers [14][15][16]. caspase-8 is another target molecule modified in a variety of cancers. Neuroblastoma and carcinomas of colorectal and head and neck tissues harbor inactivating genetic mutations of caspase-8 [17][18][19].
Epigenetic mechanisms are also implicated in the inactivation of caspase-8 during tumor development and progression [20][21]. A number of potential mechanisms have also been proposed for cancer cells to evade apoptosis through targeting the intrinsic pathway. Overexpression of the antiapoptotic BCL-2 family proteins and underexpression of proapoptotic factors, such as Bax, has been numerously reported in various cancers [22][23]. This is the predominant mechanism for the escape of cancer cells from apoptosis. Limiting the release of cytochrome C is another method to circumvent programmed cell death during cancer development. Neuroglobin (NGB) is an oxygen-binding globin protein that forms a complex with cytochrome C, preventing its release into the cytosol and activation of the caspase 9 [24]. NGB has been reported to be overexpressed in cancer cells rendering them chemo- and radiotherapy resistance [25]. Apaf-1 is another target with altered expression or activity in some types of cancer cells. These alterations have been described to block apoptosis cascade and give the ability to a cancer cell to survive and resist treatment [26][27]. Aberrant expression or function of IAP proteins is also a way used by cancer cells to avoid programmed cell death. For example, the X-linked inhibitor of apoptosis protein (XIAP) is frequently upregulated in many cancer tissues and cells and has been recognized to be responsible for the cancer cell’s resistance to various apoptotic stimuli [28].





