1000/1000
Hot
Most Recent
 +1 point
+1 point
Sarcoglycanopathies are autosomal recessive disorders and are considered rare diseases as the worldwide reported frequency is around 1/200,000–1/350,000.
Within the LGMDs, sarcoglycanopathies show particularly prominent differences among muscles. This group results from mutations in one of the four genes encoding for the transmembrane proteins of the sarcoglycan (SG) complex (α-, γ-, δ-, and β-sarcoglycan) [1][2]. All sarcoglycanopathies are autosomal recessive disorders and are considered rare diseases as the worldwide reported frequency is around 1/200,000–1/350,000 [3][4]. Additionally, two other sarcoglycans, SGCE and SGCZ, are present in the smooth muscle SG complex, but mutations in their genes have not been linked to any skeletal muscle disease yet [5]. The formation of the SG complex begins in the endoplasmic reticulum with the recruitment of the beta-sarcoglycan subunit [6], followed by the gamma, delta, and alpha subunit incorporation into the complex. Importantly, the loss of any of these proteins leads to a deficiency of the whole complex [7].
The role of the SG complex has not been fully elucidated [7][8][9]. As part of the dystrophin-associated glycoprotein complex (DAGC) that attaches the cytoskeleton to the extracellular matrix, the complex preserves the membrane stability against contraction forces [10][11] (Figure 1). Moreover, it might participate in unknown molecular pathways involved in muscle maintenance, since the loss of sarcoglycans results in muscle degeneration even without mechanical injury [12][13]. Furthermore, the SG complex is expressed in both the nervous and muscular system; yet it is only the latter that is affected in these diseases [14].
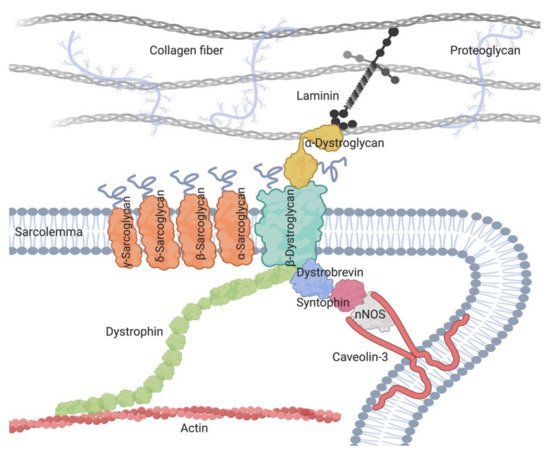
Figure 1. A schematic presentation of the dystrophin-associated glycoprotein complex (DAGC), including its main components and its localization around cell sarcolemma.
Clinically, sarcoglycanopathy patients present common traits such as elevated serum levels of creatine kinase (CK), variable muscle weaknesses (predominantly in the proximal muscles), calf hypertrophy, difficulties in arising from the floor, and the progressive loss of ambulation [15]. In the last stages of these diseases, up to 25% of patients need ventilator support [5] and cardiac involvement is present in as many as fifty percent of the cases, being more frequent in beta and gamma-sarcoglycanopathy [16][17]. However, despite their common traits, patients with sarcoglycanopathies have a very heterogeneous phenotypic profile ranging from severe Duchenne-like phenotype to mild myopathic changes [1][18][19][20] suggesting a weak correlation between genotype and phenotype. Indeed, within a large cohort of Chinese patients, a correct prediction of genotype based on the expression of sarcoglycan proteins was achieved in only 36.0% of patients [21]. Conversely, another study in alpha-sarcoglycan patients described a correlation between the residual expression of this protein and the severity of the disease [5]. No correlation for beta or gamma-sarcoglycanopathy patients could be found in the same study even though a combination of risk factors associated with rapid disease progression in these patients was identified. The low predictive power of gene mutations suggests that, in addition to gene mutations, phenotypic heterogeneity likely results from a range of other indirect and so far unknown factors.
Currently, different strategies have been proposed to prevent muscle degeneration in sarcoglycanopathies, including cell therapy [22], anti-inflammatory treatments [23], or gene therapy [24][25]; however, there is still no available cure.
Skeletal muscle is composed of contractile and non-contractile tissues. The contractile component, which is the most studied and relevant in terms of mass, consists of a variety of functionally diverse muscle fiber types [26]. One simplified classification based on their contraction and metabolic characteristics includes slow twitch oxidative and fast twitch glycolytic fibers. This fiber type classification distinguishes four main types that can be easily identified using histochemical staining techniques based on metabolic enzyme activity, such as succinate dehydrogenase (SDH) or nicotinamide adenine dinucleotide (NADH) [27]. The relative abundance of any of these fiber types within the muscle may vary between species and anatomical sites giving place to muscle heterogeneity. This heterogeneity depends on the myosin variant expressed, which might result in contractile activity features. As many as 16 different sarcomeric myosin isoforms have been described so far [26]. Some of these myosins are exclusively expressed in certain muscles. For example, jaw muscles express myosin heavy chain 16 (MYH16) that cannot be found in the trunk and limb skeletal muscles. Another source of heterogeneity can be of embryological origin. Interestingly, head muscles do not derive from somites as trunk and limb muscles do, but from presomitic cranial mesoderm [28]. However, muscle fiber heterogeneity is not restricted to myofibrillar proteins [29], metabolic enzymes [27], or developmental origin [30][31][32], but it involves also subcellular systems, including transmembrane ionic fluxes and intracellular calcium signaling [26]. These aspects require more detailed study to further understand the varying degree of disease severity among different muscles in sarcoglycanopathies.
In addition to the contractile muscle fibers, a large variety of non-contractile components contribute to the muscle organization and function, and might also contribute to muscle diversity. Among these, the connective tissues, forming tendons, ligaments, the three fascia layers (endo-, peri-, and epimysium), and the extracellular matrix (ECM) in general strongly influence muscle function [33]. Nonetheless, after acute muscle injury, transient extracellular matrix (ECM) remodeling is essential for normal muscle repair [34]. During disease, an aberrant ECM remodeling process leads to the perpetual stiffening of the ECM and ultimately creates a barrier that impairs skeletal muscle regeneration, giving rise to the most typical muscular dystrophy hallmarks: fibrosis and fat deposition [35].
Collagen is the major structural protein of these non-contractile components, accounting for 1–10% of muscle dry weight [36]. At least 28 isoforms of collagen exist across the body; among them fibrillar types I and III predominate in the adult endo-, peri-, and epimysium [37]. It is interesting that the perimysium and tendon both contain primarily type I collagen, and decorin, as the most abundant proteoglycan in both structures [33]. In contrast, epimysium and endomysium contain almost equal amounts of types I and III collagen and proteoglicans other than decorin. This fact supports the hypothesis that the perimysium is in continuity with tendons and that is very relevant due to two different factor. First, that the perimysium is the space where ECM changes mostly take place during muscle repair and during disease [38], and second, that differences among muscle-tendon arrangements have been observed along the body. For example, short fascicles are associated with long thin tendons promoting elastic energy saving, whereas long fascicles are associated with little or no tendons, favoring the ability of a muscle to produce mechanical power and control length and position [39]. These observations suggest that tendons, and the perimysium, may play a role in the development of diverse muscle phenotypes, which could have direct implications in how we approach sarcoglycanopathies.
Thus, even though the structure and function of connective tissues have been studied for some muscles [40][41], little is known about the heterogeneity within these non-contractile tissues, and even less about the morpho-functional features within the transition areas (e.g., from tendon to perimysium), especially because of the difficulty associated with isolating these “regions”.
Indeed, it is likely that some degree of diversity of connective tissues exists across the body, and it may have a potential role in muscle disease modulation.
Another factor that may contribute to muscle diversity is the heterogeneity of cell populations within the muscle. Indeed, muscle homeostasis is maintained through the cooperative actions of a variety of cell populations [42]. Among these populations, muscle stem cells, otherwise known as satellite cells [43], are responsible for myofiber growth upon activation and fusion. In addition to satellite cells, other mesenchymal stem cells found within the muscle, such as adult bone marrow derived cells [44], fibroadipogenic progenitors (FAPs) [45], mesoangioblasts [46], pericytes [47], and PW1+ interstitial cells (PICs) [48] can contribute to muscle growth and regeneration. Furthermore, during these processes, the activity of stem cells is supported by various resident and infiltrating immune cells [38][49], which help the fibers to regenerate following acute damage or in preparing the local environment for the growth and deposition of ECM during disease. Indeed, the activity of the immune system is necessary to counteract muscle degeneration in a mouse model of LGMD-R4 [50]. Another important issue to be considered for understanding these interactions in sarcoglycanopathies, as well as in other muscular dystrophies, is the regulation of the migration abilities of the different cell populations which might represent an important modulation factor [51][52].
Previous studies have described the heterogeneity of some stem cell populations, especially satellite cells. The heterogeneity of satellite cells has been described in [53], and is reflected by gene expression profiles or cell surface markers [54][55][56], as well as by their differentiation capabilities [57][58]. Likewise, other interstitial cell populations such as FAPs, isolated in mouse as lin− Sca1+, showed different levels of specific markers, such as Vcam1 and Tie2, depending on the context of the damage; acute or chronic injury [59]. Thus, single-cell gene expression profiling of skeletal muscle-derived cells has emerged as a very useful tool [60]. In addition, the work by Giordani and colleagues [61] took advantage of mass cytometry and transcriptomic profiling to reveal understudied muscle cell populations, thereby adding further complexity and highlighting the importance of uncovering new (sub)populations. Similarly, in the context of sarcoglycanopathies, Camps and colleagues [62] used single-cell technology to reveal, once again, an extensive heterogeneity among interstitial muscle cells. In particular, this study reports differences in adipogenic interstitial cells directly related to dystrophic muscles.
These interesting discoveries have raised the question as to whether additional cell subpopulations or different arrays of the already identified cell populations might be involved in muscle diversity and gradient. Indeed, we wonder whether an eventual gradient distribution of these cells may be conditioned by this context. Hence, since not all muscles respond equally to the same mutation, we believe that mouse models of sarcoglycanopathies might represent an extremely important source of different contexts in which to investigate cell population heterogeneity and their network interactions.
Muscles are apparently well-defined structures delimitated by their connective layers. However, due to their heterogeneity, it is likely that they are characterized by a gradient flow rather than as homogeneous single structures. In sarcoglycanopathies, these well-defined structures were clearly described in the work of Tasca and colleagues [63]. They showed unequivocally that muscles behaving totally differently coexist in the same leg compartment. In fact, spared or even hypertrophied muscles such as gracilis, sartorious, and rectus femoris can be observed, in proximity to the degenerating thigh adductors and posterior thigh muscles, which are the most severely affected muscles in these diseases.
Nonetheless, in the same study, considering the whole-body tissue musculature, these diverse muscles that seem to work as delimitated defined structures showed an evident proximo-distal gradient [63], recognized by the gradual change in disease severity from proximal to distal parts. This idea was supported by the observation that the distal leg compartment displays a normal or relatively preserved MRI, whereas the proximal limb muscles show a degenerated pattern. Indeed, this gradient was observed also within the same muscles, such as the quadriceps, where the distal part was spared while the proximal part degenerated.
Interestingly, the idea of a gradient along the musculature becomes more apparent when the body is studied from an architectural and geometrical point of view [39][64][65][66]. Previous studies have demonstrated a proximo-distal gradient in limb muscles as the reflection of the best compromise between energy saving, function, and architecture. Proximal limb muscles, such as the hip extensors, have long, parallel fibers that provide mechanical power, fast contraction, and precise control of joint positions to overcome gravity and move the body’s center of mass during locomotion. On the other hand, muscles with smaller mass, shorter fibers, higher pennation angles, and long compliant tendons are generally located distally within terrestrial limbs [39][64]. The collagen concentration also contributes to the above gradient, since the proximal part of the limbs contains less collagen than the distal [65].
In sarcoglycanopathies, the existence of these proximo-distal gradients reflected both in muscle pathological features and in the musculo-tendinous architecture suggest a possible unexplored relationship between the non-contractile elements such as the tendon-perimysium and muscle resistance (Figure 2).
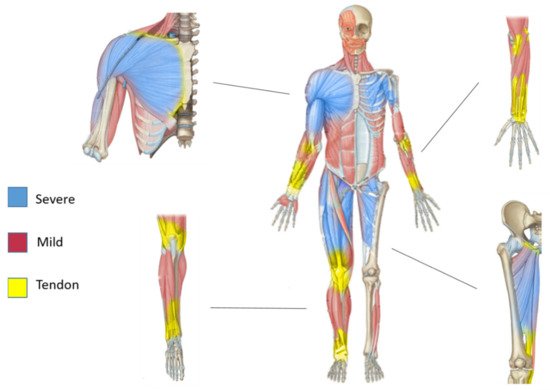
Figure 2. Proximo-distal pattern of muscles affected in sarcoglycanopathies and proximo-distal pattern in tendon architecture. Schematic presentation of human sarcoglycanopathy-affected muscle pattern with the magnification of Scheme.





