1. Introduction
Epigenetic silencing of tumor suppressor genes (TSGs) is considered a main mechanism driving cancer initiation and progression
[1][2][3]. Consequently, the reversibility of epigenetic changes has attracted attention and highlighted these changes as interesting targets in the prevention and treatment of cancer. The main epigenetic mechanisms controlling gene expression are DNA methylation and histone post-translational modifications (especially histone deacetylation and methylation), as well as the production of noncoding RNAs. The epigenetic marks left by these modifications are catalyzed by different enzymes, which can act either as writers or erasers
[4]. These enzymes work in coordination with another group of epigenetic players, called readers, which are proteins containing specialized domains that can identify and interpret an epigenetic mark in the chromatin structure and can recruit the right writer or eraser to its correct position
[4]. One intriguing epigenetic reader is a ubiquitin-like containing plant homeodomain (PHD) and an interesting new gene (RING) finger domains 1 (UHRF1), which can act as a sensor of both types of epigenetic marks (DNA methylation and histone marks) and recruit the corresponding writers, DNA methyltransferase 1 (DNMT1) and G9A, or eraser histone deacetylase 1 (HDAC1), to the right place to catalyze the same epigenetic mark
[5][6][7][8][9][10] ().
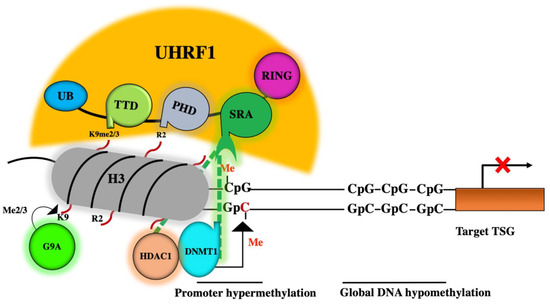
Figure 1. Role of the epigenetic reader UHRF1 (ubiquitin-like containing plant homeodomain (PHD) and interesting new gene (RING) finger domains 1) in epigenetic silencing of tumor suppressor genes (TSGs). During DNA replication, the SET and RING-associated (SRA) domain of UHRF1 can read methylated CpG sites (hemimethylated DNA) located with TSG promoter. Via the SRA domain, UHRF1 also recruits DNA methyltransferase 1 (DNMT1) and guides it to methylate the unmethylated cytosine of the newly synthetized DNA strand, leading to hypermethylation of the TSG promoter with a global hypomethylation. Through the plant homeodomain (PHD) domain, UHRF1 can bind to unmodified arginine 2 of histone 3 and via its tandem Tudor domain (TTD) domain, UHRF1 can recognize and bind to di or trimethylation of lysine 9 of histone 3 (H3K9me2 or H3K9me3). UHRF1 also uses its SRA domain to recruit histone deacetylase 1 (HDAC1) and recruits histone methyltransferase G9a, leading to histone 3 deacetylation and methylation, respectively. The consequence is the epigenetic silencing of TSGs.
UHRF1 is an oncogene that is highly expressed in several blood malignancies and solid tumors
[11][12][13][14]. It belongs to a large protein complex called the Epigenetic Code Replication Machinery “ECREM” ()
[12], which is formed through interactions between the different five domains of UHRF1 and several epigenetic writers and erasers ()
[11][12]. DNMT1, Tat Interacting Protein 60 (Tip60), a histone acetyltransferase, and the histone-lysine N-methyltransferases G9a and Suv39H1, are examples of the epigenetic writers, whereas HDAC1 and herpesvirus-associated ubiquitin-specific protease (HAUSP) serve as epigenetic erasers. The ability of UHRF1 to bind to DNMT1
[15][16][17], HDAC1
[18], Tip60
[19][20] and G9a
[21], allows UHRF1 to serve as the master conductor for connecting DNA methylation to histone epigenetic markers () and consequently ensuring their inheritance through cell division
[11][12][22]. Through these coordinated interactions, UHRF1 ensures a strong crosstalk between DNA methylation and histone post-transcriptional modifications (especially histone deacetylation and methylation), thereby silencing several TSGs, such as
p16INK4A,
hMLH1 and
BRCA1, throughout successive cell divisions, and facilitating the successful inheritance of the cancer phenotype by the daughter cells
[5][11][12]. Interestingly, several studies have reported that either UHRF1 downregulation or targeting its functional domains can act as a trigger that reactivates several TSGs and enables cancer cells to undergo apoptosis, highlighting UHRF1 as a promising target for cancer drug development
[23][24][25][26][27][28][29][30][31][32][33][34][35]. Inhibitors of UHRF1 activity and/or expression would conceivably prevent its ability to read the epigenetic markers, thereby also preventing its partners DNMT1, HDCA1 and G9a from acting out their roles as writers or erasers of epigenetic marks. The result of UHRF1 inhibition would, therefore, be the upregulation of the TSGs and subsequent activation of apoptosis pathway.
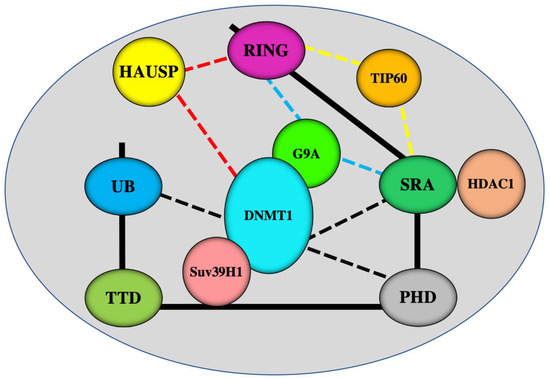
Figure 2. Schematic representation of interactions of UHRF1 domains with various epigenetic writers and erasers. UHRF1 uses its SRA domain to interact with DNMT1
[15][16][17] and HDAC1
[18]. UHRF1 can also interact with DNMT1 through PHD
[36] and ubiquitin-like domain (UBL)
[37][38] domains (black lines). HAUSP (herpes virus-associated ubiquitin-specific protease) interacts with both UHRF1 and DNMT1
[39][40] (red lines). Via its C-terminal region which covers the SRA and RING domains, UHRF1 interacts with histone methyltransferases G9a
[21] (blue lines) and histone acetyltransferase Tip60
[19][20]. UHRF1 can interacts with another histone acetyltransferase Suv39H1
[41]. DNMT1 can also interact with Suv39H1 and G9a
[42].
One interesting inhibitor of UHRF1 expression is thymoquinone (TQ), the most abundant biologically active component of black cumin seeds. Many in vitro and in vivo studies have shown that TQ exerts inhibitory effects on a number of different human cancers, including glioblastoma, breast carcinoma, leukemia, and lung, prostate, pancreatic, head and neck, cervical, and liver cancers
[43][44][45][46][47][48][49]. TQ exerts its cytotoxic activities against tumor cells by several different mechanisms, including inhibition of cell division, promotion of cell cycle arrest, activation of ROS production, induction of apoptosis and inhibition of tumor angiogenesis and metastasis
[50][51]. When compared to its effects on cancer cells, TQ has no or only mild cytotoxic effects on matched normal cells, such as normal human fibroblast cells
[52], normal human gastric epithelial cells
[53], primary normal neuronal cells
[54], normal human astrocytes
[55] and normal oral epithelial cells
[56]. Although several in vitro and in vivo studies have demonstrated the therapeutic potential of TQ as an anticancer drug for both blood malignancies and solid tumors, there is a lack of clinical studies evaluating TQ in cancer patients. This might be attributed to the poor pharmacokinetics and chemical stability of TQ. Indeed, TQ is heat and light-sensitive, and it has poor solubility in aqueous media, which affects its biodistribution
[57][58]. Additionally, covalent binding of TQ to serum albumin, and hepatic metabolism of TQ into hydroquinone, leads to significant loss of anticancer activity after oral administration
[57]. To overcome these limitations, TQ could be loaded into nanoscale systems. In this context, TQ-loaded nanoparticles have demonstrated better anticancer activity than free TQ due to enhanced bioavailability and cellular uptake
[59].
Of particular interest to this review is that several studies have now shown that TQ can target the epigenetic reader UHRF1 as well as its partners, the epigenetic writers (DNMT1 and G9A) and erasers like HDAC1
[23][60][61][62][63][64][65][66] ().
Table 1. Epigenetic targets of thymoquinone in cancer.
|
Epi-Target
|
Role of Epi-Target
|
Experimental Model
|
Mechanisms of Action
|
References
|
|
UHRF1
|
Reader
|
Human cervical carcinoma HeLa cells.
T-ALL
|
TQ targeted the E3 ubiquitin ligase activity of UHRF1 resulting in an auto-ubiquitination of UHRF1 likely through the downregulation of HAUSP
|
[23]
|
|
T-ALL
|
TQ upregulated p73 expression and cleaved caspase 3 leading to UHRF1 degradation
|
[63]
|
|
T-ALL
|
TQ decreased the expression of PDE1A leading to the upregulation of p73 and downregulation of UHRF1
|
[62]
|
|
T-ALL
Human breast cancer cells
|
TQ decreased the expression of mRNA UHRF1 in dose-dependent mechanism
|
[60]
|
|
DNMT1
DNMT3A
DNMT3B
|
Writer
|
Human acute myeloid leukemia cells
Patient primary cells
|
TQ inhibited DNMT1 activity and decreased its expression through the disruption of Sp1/NFkB complex from DNMT1 promoter.
TQ decreased the expression of DNMT3A through the upregulation of miR-29b, known to directly bind to the 3′-UTR of DNMT3A
|
[64]
|
|
T-ALL
|
TQ decreased the expression of DNMT1 protein
|
[63]
|
|
T-ALL
|
TQ decreased the expression of DNMT1, 3A,3B
|
[60]
|
|
HDAC1
HDAC2
HDAC3
HDAC4
HDAC9
|
Eraser
|
T-ALL
|
TQ decreased the expression of HDAC1 protein
|
[63]
|
|
T-ALL
|
TQ decreased in the expression of HDAC1, 4 and 9
|
[60]
|
|
T-ALL
Human breast cancer cells
|
TQ decreased the expression of mRNA HDAC1 in dose-dependent mechanism
|
[60]
|
|
Human pancreatic ductal adenocarcinoma cells.
Human pancreatic ductal adenocarcinoma xenografts.
|
TQ inhibited HDAC activity, decreased the expression of HDAC 1, 2, 3 at mRNA levels and increased the acetylation of histone 4 at lysine 12 (H4 Ac-K12)
|
[65]
|
|
G9A
|
Writer
|
T-ALL
Human breast cancer cells
|
TQ decreased the expression of mRNA G9A in dose-dependent mechanism
|
[60]
|
The TQ-induced inhibition of these epigenetic players is associated with an upregulation of several TSGs that are known to be repressed in several tumors through epigenetic mechanisms. Indeed, TQ can induce the degradation of UHRF1 through a fast autoubiquitination process involving the UHRF1 RING domain, which has a specific E3 ubiquitin ligase activity
[23]. Interestingly, UHRF1 ubiquitination was not observed in TQ-treated cells that expressed a mutant form of UHRF1 with a specifically modified RING domain, indicating that the RING domain of UHRF1 undergoes autoubiquitination in response to TQ treatment
[23]. Moreover, the deubiquitinase HAUSP, which is known to protect UHRF1 from degradation by the proteasome
[39][40], was also downregulated in response to TQ, suggesting that TQ could be the trigger for the autoubiquitination of UHRF1 by an as yet unknown mechanism
[23]. Nevertheless, it is absolutely established that the dissociation between HAUSP and UHRF1 is involved.
An effective epidrug for cancer therapy should consider the epigenetic code as a whole, rather than a single pharmacological target
[67][68]. Single epitarget therapies suffer from some significant limitations, particularly the emergence of drug resistance and the triggering of adverse reactions
[69]. Identifying compounds with multitargeting properties that are active against epigenetic marks should overcome these limitations. TQ, through its ability to target the expression of both the epigenetic reader UHRF1 and its preferred partners DNMT1, HDAC1 and G9a, is clearly a potential candidate as a multitarget epidrug with the capacity to reverse the epigenetic code of cancer cells as a whole, while allowing the reactivation of TSGs.
2. Role of the DNMT1/HDAC1/G9a Complex in Epigenetic Silencing of TSGs
Inactivation of TSGs through epigenetic mechanisms (DNA methylation and histone posttranslational modifications) is one of key factors that promotes the onset of cancer. In cancer cells, the methylation profile is characterized by a global genome hypomethylation, accompanied by a hypermethylation of TSG promoters. The hypermethylation of the CpG islands in TSG promoters, catalyzed by DNMT1, is a significant event in the origin of many cancers
[70][71]. Many TSGs, such as
RB1,
VHL,
p16INK4a,
BRCA1,
HIC-1,
MLH1, RUNX3, RASSF1A, FOXO4, PPARG, STK4, PML and
KISS1, are silenced in tumors by hypermethylation of their promoters
[1][11][12]. TSGs regulate several signaling pathways involved in cell proliferation, the cell cycle, DNA repair, invasion, apoptosis and angiogenesis, all of which are involved in the initiation and/or the development of cancer
[11][13].
Apart from creating an imbalance in DNA methylation, aberrant histone post-translational modifications can also drive the epigenetic inhibition of TSGs in cancers. Aberrant histone post-translational modifications are particularly prevalent in cancer cells
[72][73], with post-translational histone modifications usually occurring in the early stages of tumor development and accumulating during tumorigenesis
[74][75]. The post-translational modifications occurring at certain sites on histones H3 and H4 are among the most important modifications that exert effects on gene expression
[76][77][78]. This modification process is mediated by two types of enzymes with opposite activity: histone acetyltransferases (HATs) and histone deacetylases (HDACs).
The downregulation or upregulation of HATs is accompanied by tumorigenesis or poor prognosis
[79][80]. The Tip60 histone acetyltransferase can acetylate histone proteins, such as histone 2A on lysine 5 (H2AK5)
[81], as well as several nonhistone proteins, including p53 and Myc
[82], and Tip60 can serve either as an oncogene or as a tumor suppressor
[83]. Several tumors, such as skin cancer
[84] and osteosarcoma
[85], show overexpressed Tip60, whereas other tumors, including lung
[86] and breast cancer
[87], show low tumor expression levels of Tip60.
The HDACs, a family of four enzyme subclasses, also have a vital role in tumorigenesis, and are attracting attention due to their contributions to several biological processes, as well as their interactions with other epigenetic enzymes
[79]. Changes in HDACs expression in tumors usually result in aberrant deacetylation, leading to activation of TSGs. HDAC1, a class I member, is considered an important epigenetic player mediating histone deacetylation
[88][89][90]. HDAC1 is highly expressed in many human tumors, and its overexpression is associated with poor outcomes and tumor progression, thereby identifying this epigenetic eraser as a promising target for cancer therapy
[88][91][92][93]. Downregulation of HDAC1 inhibits cell proliferation and cell cycle progression and induces apoptosis in many human tumors, including breast and colon cancer cells, ovarian cancer and lung cancer
[90][94][95]. Clinically, HDAC1 overexpression at the mRNA and proteins levels is associated with the clinical features and poor prognosis of patients with breast
[96], lung
[88] and gastric cancer
[97], supporting the idea that the inhibition of HDAC1 activity and/or HDAC1 expression could be a potent strategy for cancer therapy.
Epigenetic changes involving histone methylation/demethylation are dynamically regulated by two families of enzymes: histone lysine methyltransferases (KMTs) and lysine-specific histone demethylase (KDMs)
[98]. Lysine residues can be mono, di or trimethylated, and this process is regulated by the expression levels and the recruitment of KMTs/KDMs to chromatin. The methylation of lysine within histone tails, catalyzed by KMTs, plays a central role in the control of gene transcription
[99][100]. G9a, also known as EHMT2 (euchromatic histone-lysine N-methyltransferase 2) and KMT1C (lysine methyltransferase 1C), is one of the major euchromatic methyltransferases
[101][102]. The di- or trimethylation of lysine 9 of histone 3 (H3K9me2 or H3K9me3) mediated by the KMT G9a is considered one of histone changes with a known role in gene silencing, whereas the methylation of lysine 4 (H3K4me) on the same histone is related to gene activation
[103][104]. Several studies have shown that G9a is overexpressed in a panel of human cancers, and that its high expression levels are associated with unfavorable clinicopathological parameters and poor survival
[105][106][107][108]. Interestingly, the depletion of G9a is sufficient to induce a reactivation of TSGs and inhibition of cancer cell proliferation
[109][110][111].
Several other studies have also reported a coordinated activity between several epigenetic factors, including DNMT1, HDAC1, G91 and Suv39H1, during DNA synthesis that maintains the transmission of the epigenetic code
[42][112][113][114][115][116]. In this context, DNMT1 was shown to physically interact with both the H3K9 histone methyltransferases G9a and Suv39H1
[42]. DNMT1 colocalized with G9a at replication foci during DNA replication, while DNMT1 colocalized with Suv39H1 on heterochromatic regions predominantly before cell division
[42]. The DNMT1/HDAC1/Suv39H1 complex, in coordination with other factors, was found to regulate the expression of the estrogen receptor-a (ER) in breast cancer cells
[116].
A key remaining question is whether a principal conductor exists that coordinates all these epigenetic factors to orchestrate the precise timing of the recruitment of the correct enzyme to its right place. In this review, we propose that UHRF1 is a likely candidate. Based on its structure and its multiple interactions with several writers and erasers, UHRF1 can direct coordinated crosstalk between DNA methylation and histone posttranslational modifications, thereby making it a probable candidate as the leader in the ECREM complex.
3. A master Role for UHRF1 in the ECREM Complex Driving Epigenetic Inhibition of TSGs
In tumors, no clear mechanisms are yet identified that explain the maintenance of the inheritance of a silenced TSG from a mother cancer cell to the daughter cells during cell division. However, the functioning of UHRF1 as the conductor and hub protein in its complex could ensure this transmission during cell division
[5][6][12][14][15][16][20][22][36][78]. Indeed, several in vitro and in vivo studies have reported the detection of UHRF1 overexpression in many human cancers, and that this overexpression is a crucial factor in the epigenetic silencing of various TSGs and leads to enhanced cell proliferation, cell cycle progression, and suppression of apoptosis
[11][12]. UHRF1 uses its different domain functions to repress the expression of TSGs through several mechanisms that involve TSG promoter hypermethylation via the physical interaction with DNMT1, HDAC1-mediated histone deacetylation, and G9a-catalyzed histone 3 methylation
[11][12] ().
The induction of TSGs silencing is well documented to occur by hypermethylation of CpG islands located within TSGs promoters. However, the mechanisms by which the CpG islands are specifically targeted is still unclear. One hypothesis is that the hypermethylation of the CpG islands in the TSG promoters is driven by a mechanism involving a protein that can bind to DNA and guide DNMT1 to its correct place at the right time during DNA replication. UHRF1 is a likely candidate because of its high affinity for hemimethylated vs. nonmethylated DNA
[117], and its direct interaction with DNMT1
[15][16][17][36]. These capabilities give UHRF1 the necessary duality to allow the successful transfer of DNA methylation patterns, including the hypermethylation of TSGs. During DNA replication, the SRA domain of UHRF1 can recognize methylated CpG sites (hemimethylated DNA) by flipping out the methylated cytosine. In addition, via the same domain, UHRF1 can recruit DNMT1 and guide it to methylate the unmethylated cytosine of the newly synthetized DNA strand
[117] (). UHRF1 can also interact with DNMT1 through its PHD domain
[36] and UBL domain
[37][38] (). Beside the role of SRA domain of UHRF1 in the binding to hemimethylated DNA and the recruitment of DNMT1 to sites of methylation DNA
[117], UHRF1 has a well-established role through its RING domain in the ubiquitylation of H3 and DNMT1, targeting for sites of hemimethylated DNA
[118][119]. Indeed, UHRF1 uses the ubiquitin ligase activity of its RING domain to ubiquitinate H3. The reading and writing this epigenetic mark (H3 ubiquitination) by UHRF1 is a prerequisite for the binding of DNMT1 to ubiquitylated histone H3
[120] to ensure a faithful recruitment of DNMT1 to sites of hemimethylated DNA
[22][118][119]. Moreover, the recruitment of DNMT1 to DNA methylation sites is also regulated by H3 deubiquitylation through a mechanism involves HAUSP, another member of ECREM complex
[121]. HAUSP was shown to interact with DNMT1 and is recruited to sites of DNA methylation during DNA replication, and this recruitment requires UHRF1
[121]. HAUSP induced the deubiquitylation of ubiquitylated histone H3 in vitro, while HAUSP depletion in cancer cells resulted in enhanced histone H3 ubiquitylation
[121]. This suggests that HAUSP has a key role in the regulation of maintenance of DNA methylation through UHRF1-dependent deubiquitylation of ubiquitylated histone H3. However, HAUSP and DNMT1 appear to behave as independent proteins at replication foci, since global DNA methylation levels were not notably altered in cells with HAUSP knockout
[122][123].
Several works have shown that both histone methylation and acetylation work together with DNA methylation to exert inhibitory effects on the expression of TSGs in cancer cells through mechanisms that remain incompletely understood
[124][125][126][127][128]. Through its SRA domain, UHRF1 can directly bind to HDAC1 and recruit it to methylated promoter regions of the TSGs
p16INK4A and
p14ARF, resulting in their silencing by a histone deacetylation process
[18]. In the same context, renal cell carcinoma (RCC) tumors show high expression levels of UHRF1 compared to normal renal tissues, and this overexpression is associated with a decreased expression of the tumor suppressor gene
TXNIP [129]. UHRF1 was shown to recruit HDAC1 to the
TXNIP gene promoter and mediate the deacetylation of histone H3 on the lysine 9 (H3K9), resulting in an epigenetic inhibition of TXNIP expression
[129]. Interestingly, UHRF1 downregulation in RCC cell lines induced the upregulation of TXNIP expression and apoptosis, suggesting that UHRF1 inhibits the expression of TXNIP in RCC through epigenetic mechanisms, thereby promoting tumor progression
[129].
The UHRF1/HAUSP/DNMT1 complex was also detected on the promoters of
HHIP and
IGFBP3, two key TSGs in hepatoblastoma, and this interaction caused the inhibition of these genes
[130]. Silencing of
HHIP and
IGFBP3 genes was associated with an increase in the dimethylation of histone 3 on lysine 9 (H3K9me2)
[130], which is a well-documented repression mark in cancer
[131][132]. Interestingly, the depletion of UHRF1, but not of its partner HAUSP, significantly increased the expression of the
HHIP and
IGFBP3 genes and decreased the H3K9me2 mark at the
HHIP and
IGFBP3 TSG loci, leading to the inhibition of hepatoblastoma cell growth
[130]. Similarly, UHRF1 was also shown to recruit DNMT1 to the promoter of the tumor suppressor gene
BRCA1 leading to
BRCA1 inhibition through methylation of its promoter
[133]. Besides guiding DNMT1, UHRF1 also recruited HDAC1 and G9a to the
BRCA1 loci, resulting in histone 3 deacetylation and methylation, respectively, and facilitating the silencing of
BRCA1 [133].
Taken together, the findings of these studies support the idea that UHRF1 overexpression is one of the primary causes of cancer pathogenesis, and that UHRF1 exerts direct inhibitory effects on various TSGs through a coordinated recruitment of several epigenetic players, namely DNMT1, HDAC1 and G9a, to their correct places on the chromatin to catalyze the right epigenetic mark (). These studies also reinforce the view that DNMT1, HDAC1 and G91 might coregulate the expression of TSGs in cancer, and that this process is directly under the control of the epigenetic reader UHRF1 (). Thus, understanding the role of the UHRF1/DNMT1/HDAC1/G9a complex in reading the epigenetic marks (DNA methylation and histone marks) in cancer will allow the development of a new generation of multitarget epidrugs. These drug candidates may have value in targeting UHRF1 as the principal conductor, with subsequent regulatory effects on the other partners, DNMT1, HDAC1 and G91.
 +1 point
+1 point