1. Diagnosis
The diagnostic criteria for CLL, although refined over time, have not dramatically changed since the first guidelines stablished in the 1990s [
17,
18]. In 2008, the International Workshop on Chronic Lymphocytic Leukemia (iwCLL) published consensus guidelines with updated recommendations for the management of CLL in general practice [
19]. This version was updated with minor modifications in terms of the diagnostic criteria in 2018 [
20]. Nowadays, the diagnosis of CLL is mainly based on laboratory features, namely blood count, morphology and immunophenotyping [
1,
20].
CLL is first suspected when an absolute peripheral lymphocytosis of 5 × 10
9/L clonal B cells is found in the peripheral blood [
17]. This lymphocytosis must persist for longer than 3 months, according to the latest version of iwCLL guidelines [
20]. The presence of a cytopenia caused by clonal bone marrow involvement establishes the diagnosis of CLL regardless of the peripheral B-lymphocyte count [
20]. Bone marrow aspirate and biopsy are not required for the diagnosis of CLL. However, if done, the marrow often demonstrates >30% lymphocytes [
21].
The leukemia cells in the blood smear are characteristically small, mature lymphocytes with a narrow border of cytoplasm and a dense nucleus lacking discernible nucleoli and having partially aggregated chromatin. Large atypical cells, cleaved cells and prolymphocytes are also often seen on the peripheral smear and may account for up to 55% of the peripheral lymphocytes [
17,
22].
The clonality of the peripheral circulating B-lymphocytes needs to be confirmed by flow cytometry. Based on the antigenic profile, Matutes et al. designed in 1994 an immunologic score system (Matutes score, MS) to ensure the diagnosis of CLL [
23]. In this scoring system, a value of 0 or 1 was given according to the expression of the five markers of CD5, CD23, FMC7, surface immunoglobulin M (sIgM) and CD22. Most CLL cases had a score of 4 or 5, whereas non-CLL cases had a score of less than 4. It was shown subsequently that CD22 could be advantageously replaced by CD79b [
24]. The scoring proposed in the modified MS has been the basis of diagnosis for the following years and was defined by a strong expression of CD5 (normally expressed on T cells) and CD23, a low or absent expression of CD79b, sIgM and FMC7 [
24]. However, in some cases, differential diagnosis on the basis of the markers included in this score has been challenging due to some limitations affecting reproducibility—in particular, flexibility in marker expression.
Other potentially informative markers have been evaluated to be considered for the CLL diagnosis, although a consensus concerning these novel markers has not been reached yet [
25,
26,
27,
28,
29]. In 2018, a recent large harmonization effort confirmed that a panel of CD19, CD5, CD20, CD23 and sIg kappa or lambda is usually sufficient to establish the diagnosis of CLL using peripheral blood samples [
30]. In borderline cases, markers such as CD43, CD79b, CD81, CD200, CD10 or ROR1 may help to refine the diagnosis [
30]. The current criteria for the CLL diagnosis have been updated by the iwCLL, the World Health Organization (WHO) and the European Research Initiative on CLL (ERIC) [
8,
20,
30,
31]. However, MS is still used in many centers. A lymphoid node biopsy and/or bone marrow biopsy may be helpful if immunophenotyping is not conclusive for the diagnosis of CLL [
2].
The 2008 WHO classification included CLL, together with small lymphocytic lymphoma (SLL), as mature B-cell neoplasms entities [
32]. SLL is characterized by the presence of fewer than 5 × 10
9/L lymphocytes with lymphadenopathy and without cytopenias, although this diagnosis should be confirmed by a lymph node biopsy.
The 2008 WHO classification of lymphoid neoplasms also defined MBL when the presence of less than 5 × 10
9/L clonal B lymphocytes happened in the absence of lymphadenopathy or organomegaly (as defined by a physical examination or CT scan), cytopenias or disease-related symptoms [
32,
33,
34,
35]. About 1% to 2% of MBL cases progress to CLL per year [
36]. In the 2016 update, the WHO differentiated “low-count MBL” from “high-count MBL” according to the size of the monoclonal B-cell population (cutoff: 0.5 × 10
9/L) [
8].
As previously mentioned, CLL can develop Richter Syndrome (RS), a secondary and aggressive lymphoma with an incidence rate of ~0.5% per year. Most cases of RS (95%) consist of a histologic transformation to diffuse large B-cell lymphoma (DLBCL) and, less often, Hodgkin’s lymphoma (HL) [
37]. Commonly, RS clinically presents with rapidly enlarging lymph nodes, accompanied by the presence of constitutional symptoms, fever and weight loss, together with elevated lactate dehydrogenase (LDH) levels, when lymph nodes enlarge rapidly [
9,
38]. A lymph node biopsy is required to establish the diagnosis of a transformation into an aggressive lymphoma [
19]. DLBCL-RS is clonally related to the underlying CLL in more than 80% of cases and has a worse outcome than clonally unrelated cases, which have a prognosis similar to de novo DLBCL [
39].
Once the diagnosis of CLL is confirmed, patients should undergo additional laboratory evaluations to help the physician predict the prognosis and guide the treatment approach. Very recently, the ESMO Clinical Practice Guidelines provided recommendations on the management of CLL for diagnosis, treatment and follow-up [
2].
2. Treatment
2.1. Treatment Evolution on the Last Decades
Advances in the understanding of CLL biology have resulted in the development of new therapeutic approaches that have dramatically improved patient outcomes [
186]. Recently, the identification of the specific therapeutic targets involved in the intracellular signaling pathways, such as the B-cell receptor (BCR) or BCL-2 (B-cell lymphoma), has revolutionized the treatment of CLL patients. CIT-based regiments were the standard of care for many years but have taken a backseat, with TA and their combinations occupying first place due to their excellent efficacies. The development of second-generation anti-CD20 molecules, in combination with targeted molecules, has also contributed to the changes in the therapeutic landscape. Although the majority of CLL patients with an active disease have benefited from this progress, probably those with major improvements in their quality of life and life expectancy have been elderly and/or high-risk patients [
187]. However, the new treatment approaches also come with challenges, such as the emergence of drug resistance, toxic and adverse effects and treatment costs. Combination therapies, as well as the incorporation of other TA, will help to optimize the treatment approaches in the near future.
Other approaches, such as radiation therapy or splenectomy, have been abandoned in favor of CIT or TA, in most cases [
188]. An exception in which these treatments might be considered is in a palliative setting. As CLL lymphocytes are radiation-sensitive, radiotherapy might be used in a palliative patient with compression symptoms [
189]. A splenectomy might be effective for patients with massive splenomegaly refractory to other treatments [
190]. Despite the great treatment evolution during the last years, it is important to point out that the majority of CLL patients are still monitored with a ‘watch and wait’ approach until the balance of risks and benefits favors the treatment initiation [
191]. Indeed, a substantial fraction of CLL patients do not require CLL-related therapy during their lifetime [
7].
2.1.1. Chemoimmunotherapy
Over the past 50 years, and before the introduction of TA, the activity of the chemotherapy agents comprising alkylating agents (chlorambucil, cyclophosphamide and bendamustine); nucleoside analogs (fludarabine, pentostatin and cladribine) and corticosteroids was remarkable in patients with CLL. At the beginning, chlorambucil monotherapy was the therapeutic “gold standard” for several decades, but later, fludarabine-based regimens took advantage due to their superior overall response rates (ORR) compared with the other treatment regimens containing alkylating agents or corticosteroids [
192].
In the early 2000s, the addition of anti-CD20 antibodies to chemotherapy resulted in prolonged survival, and CIT regimens therefore became the gold standard therapy. The combination of fludarabine, cyclophosphamide and rituximab (FCR) [
193,
194] was commonly used for younger, fit patients; bendamustine combined with rituximab (BR) [
195,
196,
197] was commonly used for unfit patients and chlorambucil with anti-CD20 antibodies was used for elderly patients with coexisting conditions [
198,
199]. One of the potential risks of anti-CD20 antibodies is the reactivation of hepatitis B. Thus, virus B serologic testing is mandatory in all patients before anti-CD20 treatment initiation, and prophylactic antiviral therapy must be initiated before treatment in cases with a risk of reactivation. Another worrying issue associated with CIT is the long-term risk of inducing secondary neoplasia, including myelodysplastic syndromes and acute myeloid leukemia [
61].
In the last years, some randomized clinical trials improved the survival and showed better side effect profiles with the TA [
200,
201]. Nowadays, the use of chemoimmunotherapy is steadily declining. An exception could be the group of young fit patients with
IGHV-M, who often stay in remission for more than 10 years after treatment with the FCR regimen. For such patients, FCR treatment remains an alternative to the TA until we have a longer follow-up on ibrutinib-treated patients [
61].
2.1.2. Bruton Kinase Inhibitors
Ibrutinib is an oral small molecule acting as a Bruton tyrosine kinase inhibitor (BTKi). This drug is widely used nowadays not only as a frontline treatment but, also, in the relapse setting [
113,
202]. This is supported by the very satisfactory results recently shown in the phase 3 clinical trials RESONATE [
203] and RESONATE-2 [
204]. Even though, in both trials, the control arm was not the best “standard of care”, their results were impressive, showing a high ORR and survival benefit in the ibrutinib arm, with a follow-up of more than 5 years, for all the CLL subgroups. The first results from RESONATE showed the excellent efficacy of ibrutinib in refractory/relapse (R/R) CLL patients, leading to Food and Drug Administration (FDA) approval in 2014 [
205]. The second clinical trial experimented the use of ibrutinib as a frontline therapy [
204]. More recently, ibrutinib was compared to CIT in treatment-naive CLL patients, questioning the need for CIT even in the subgroup of young, low-risk patients. The combination of ibrutinib–rituximab (IR) was superior to FCR in terms of the PFS and OS in ECOG-ACRIN E1912. This benefit was observed for all the analyzed subgroups, with the exception of
IGHV-M patients, in which both treatments achieved similar results, and a long follow-up is required to determine the best option for this population [
201]. For patients not able to tolerate FCR, the ALLIANCE trial compared BR to ibrutinib +/− rituximab. Patients receiving ibrutinib showed a longer PFS than patients treated with BR. Benefits in the OS have not been observed to date, with a median follow-up of 38 months. Furthermore, rituximab did not improve the PFS compared to patients treated with ibrutinib monotherapy [
200].
Ibrutinib is not free from adverse events, with the most frequent being mild diarrhea, fatigue, nausea, bruising and arthralgia, while the most severe and less common are infections, atrial fibrillation, hypertension and ventricular arrhythmia [
210]. Additionally, the data from real-life studies show that the major cause of discontinuation is off-target toxicity rather than progression [
211]. Probably, a better selection of patients with cardiovascular comorbidities or at a high risk of bleeding or infection can optimize this discontinuation rate. On the other hand, potential benefits of ibrutinib include a modulating effect on the immune system [
212].
Currently, second-generation BTKi are under investigation. These inhibitors join more selectively to their therapeutic target, improving their toxicity profile due to less frequency of the off-target events. Of them, acalabrutinib is the most mature, as the FDA has recently approved it for CLL patients (first-line and relapse) based on last year’s results of the ELEVATE-TN [
206] and ASCEND [
207] phase 3 clinical trials. Both trials demonstrated superiority in the acalabrutinib arms, with a good safety profile, as shown in . Moreover, the addition of obinutuzumab to acalabrutinib could provide a better PFS than acalabrutinib monotherapy in therapy-naive CLL patients but neutropenia in 30% of patients [
206]. Zanubrutinib or tirabrutinib are the other second-generation BKTi under clinical development. Both have demonstrated encouraging activity in CLL patients, with a low incidence of off-target toxicity in their phase 1 and 2 studies [
213,
214]. Specifically designed to overcome the acquired resistance to ibrutinib, a new family of reversible BTKi emerged in 2020. These agents are now in the early phases of research and are soon to demonstrate their applicability in real life. Among them, fenebrutinib, LOXO 305 and ARQ 531 are under active clinical investigation nowadays [
215].
2.1.3. BCL-2 Inhibitors
Venetoclax is an oral BCL-2 inhibitor highly active in patients with CLL. The clinical development of this drug has lagged behind that of ibrutinib, although its effectiveness seems just as promising. The first clinical trials of venetoclax in patients with R/R CLL showed high response rates in terms of the PFS and ORR across all subgroups of CLL patients [
216,
217]. Based on the CLL-14 and MURANO phase 3 trials () [
208,
209,
218,
219], venetoclax in combination with anti-CD20 has been recently approved for frontline treatment and treatment for relapsed CLL. As opposed to BCR pathway inhibitors, venetoclax induces deep remissions with high rates of MRD that allow treatment discontinuation. To date, venetoclax plus obinutuzumab has yielded the highest MRD-negative response rate in a randomized trial so far [
220]. An extended follow-up of the CLL-14 and MURANO trials has recently been published, confirming the notorious clinical benefit of the combinations with venetoclax and demonstrating an OS benefit for R/R patients treated with venetoclax–rituximab. The rates of MRD negativity were also significantly higher in the venetoclax arm of both trials [
208,
218].
Venetoclax requires special measures (initial ramp-up escalation dose, vigorous hydration and laboratory test monitoring) to mitigate the risk of tumoral lysis syndrome (TLS) observed in the first clinical studies. Taking into account the aforementioned factors, TLS is not a big concern and has been reported in a low proportion of patients. In contrast, the most frequent grade ≥3 adverse event is neutropenia detected in around 50–60% of the patients, although not followed by a higher risk of infection [
208,
218].
2.1.4. PI3K Inhibitors
Idelalisib is the first-in-class phosphatidyl-inositol 3-kinase inhibitor (PI3Ki) used in R/R CLL patients. Its clinical development was contemporary to ibrutinib, and it has been demonstrated to be an active oral small molecule, preferably used in combination with rituximab. This was shown in a phase 3 study that randomized 220 patients to receive rituximab plus idelalisib or a placebo. Patients receiving idelalisib significantly improved their PFS (19 vs. 6 months) and their OS (41 vs. 35 months), despite an extensive cross-over [
221], and achieved a high rate of ORR. However, these benefits seem inferior to those obtained with BTKi, as was recently confirmed in the ASCEND clinical trial [
207].
Toxicity has limited the use of idelalisib in real life, with a high rate of infectious (pneumonia) and autoimmune side effects (colitis, pneumonitis and hepatitis). Duvelisib is another PI3Ki granted by the FDA in 2018 for the treatment of R/R CLL patients after at least two prior therapies. It has also been demonstrated to be active in CLL, but, again, toxicity might limit its widespread use. Umbralisib is a next-generation PI3Ki with a much better toxicity profile, as it has been related to fewer immune-mediated toxicities or severe opportunistic infections to date [
222]. Different clinical trials of umbralisib alone or in combination are ongoing and will help to elucidate its role in this rapidly changing treatment era.
2.1.5. Immunotherapy
The surface antigen CD20 is the target of antibodies such as rituximab, ofatumumab and obinutuzumab, which are currently approved for CLL. These antibodies are commonly administered in combination with chemotherapy or targeted therapies.
Recently, advances in monoclonal antibody technology have resulted in the development of new antibodies with improved therapeutic effectiveness. Ublituximab stands out, a next-generation CD20 antibody with encouraging results, especially in combination with the targeted molecules [
222] or other monoclonal antibodies such as cirmtuzumab (anti-ROR 1), MOR00208 (anti-CD19) or otlertuzumab (anti-CD37). Less advanced are the bispecific antibodies and immunomodulatory antibodies [
215].
Alemtuzumab, as anti-CD52 monoclonal antibody, is approved for the treatment of CLL. It was indicated, before the TA “era”, especially for patients with del(17p)/
TP53 mutations. However, the use of alemtuzumab is exceptional today due to the severe immunosuppression and the high rates of infectious complications associated with this drug [
223]. Moxetumomab pasudotox, an antibody–drug conjugate targeting CD22 and delivering a cytotoxic agent simultaneously, has also been unsuccessful in treating CLL, unlike hairy cell leukemia. This is explained by the lower expression of CD22 in CLL lymphocytes [
224].
2.1.6. Combinations of Novel Agents
TA have changed the treatment landscape of CLL. Combinations of these targeted treatments with CIT, CD20 monoclonal antibodies and between them is what the immediate future holds. This approach aims to limit the toxicity, cost and resistance and achieve profound responses with MRD that can lead to the potential curation of the disease and treatment discontinuation. In particular, existing evidence indicates that anti-CD20 plays a synergistic role when used in combination with venetoclax. The combination of second-generation anti-CD20, such as obinutuzumab with BTKi, also appears to be beneficial [
196,
218,
220,
225].
Regarding the combinations between CIT and TA, probably the most interesting studies are those including young, fit, treatment naïve patients with mutated IGHV. Some of them have demonstrated very high rates of negative MRD, allowing the discontinuation of TA. Ultimately, combinations between TA with or without the addition of anti-CD20 have shown preliminary promising outcomes, with high rates of MRD negativity making possible treatment discontinuation as well. Nowadays, countless clinical trials are ongoing on this field.
Moreover, three ongoing, independent phase 3 trials stand out (ECOG-ACRIN EA9161 for young patients, ALLIANCE A041702 for patients >70 years old and CLL-17 (fit and unfit patients)), exploring different combinations with venetoclax, ibrutinib and obinutuzumab that allow treatment disruption in some of their arms. The results of these studies will probably again change routine practices in the near future.
2.1.7. Cellular Therapy
Allogeneic stem cell transplantation (allo-TPH) is a potentially curative approach to CLL patients. Years ago, it was indicated in patients with poor prognostic factors (early relapses, refractory to fludarabine or harboring TP53 abnormalities). TA have changed the natural history of CLL, and therefore, the role of allo-TPH in this new era is less clear. A recently published retrospective study reported the outcome of 65 patients undergoing allo-TPH after at least one TA, pointing out that it is a viable long-term disease control strategy. In this study, the investigators observed that PFS was predicted by the hematopoietic cell transplantation-specific comorbidity index. No differences were observed among the patients receiving previous TA (one or two ibrutinib/venetoclax) or TA and CIT as the previous treatment [
235]. Currently, most guidelines recommend it for patients with high-risk CLL that have relapsed or are refractory to at least one TA or in cases of clonally related Richter transformation with a response to chemotherapy [
202,
236]. However, some questions, such as the optimal timing of the procedure, remain unanswered.
CLL was a pioneering disease in which chimeric antigenic receptor T (CAR-T) cells targeting CD19 were tested [
237], but the ORRs were not as good as those observed in other diseases, and the estimated PFS at 18 months was around 28% [
238]. In order to optimize its applicability in CLL, different strategies are under investigation, such as those using ibrutinib concurrently with CD19 CAR T cells [
239]. With this approach, the ORR and PFS were improved, and a better toxicity profile was observed after one year of follow-up. Even so, these results need more robustness to be adopted in clinical practice. Another option under study is the use of modified cord blood natural killer cells to express anti-CD19 CAR [
240].
2.2. Current Treatment Strategies
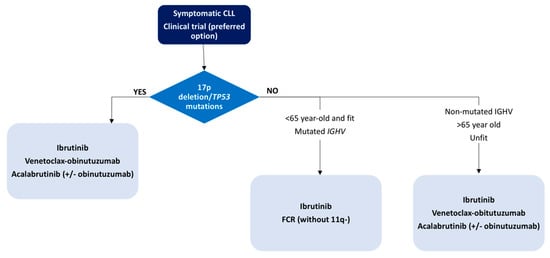
In contrast to the treatment paradigm shift previously described, the treatment indications remain without changes, as outlined by the consensus guidelines published by the iwCLL in 2018 [
20]. For the time being, asymptomatic patients must be monitored without active treatment irrespective of the risk, even though some studies treating high-risk asymptomatic patients are ongoing, aiming to answer if this approach is beneficial [
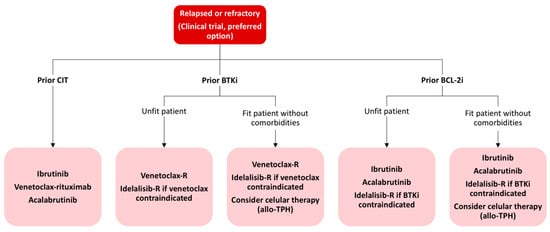
241]. With the existing evidence and actual approval, we propose a treatment algorithm based on patient age, comorbidities and genetic abnormalities, as depicted in and .
Figure 2. First-line treatment algorithm for CLL patients.
Figure 3. Treatment algorithm for relapsed or refractory CLL patients. CIT: chemoimmunotherapy; BTKi: Bruton tyrosine kinase inhibitor; R: rituximab; allo-TPH: allogeneic stem cell transplantation; BCL-2i: BLC2 inhibitor.
2.3. Drug Resistance
Despite the significant clinical efficacy in most CLL patients treated with TA, in some of them, the treatment fails. The number of patients who progress or develop clinical resistance is expected to increase in the following years due to the increasing number of patients treated with TA and the long-term administration of these agents. Thus, understanding the potential resistance mechanisms will help to design new treatment strategies to prevent resistance and avoid relapse.
2.3.1. Ibrutinib Resistance
While ibrutinib is an effective therapy leading to durable responses, some patients acquire resistance and relapse [
242]. In 2014, a study using whole-exome sequencing discovered acquired mutations within the
BTK gene in CLL patients relapsing on ibrutinib [
243]. Further studies confirmed the presence of a
BTK mutation in the CLL patients relapsing on ibrutinib [
115,
244,
245], C481S being the most common mutation at the position of the binding site of the drug [
246,
247].
BTK mutations can be explained by the mechanism of action of ibrutinib, which binds to BTK with an irreversible covalent bond at position C481S. From there, ibrutinib inhibits the proliferative and antiapoptotic signals that are abnormally stimulated in CLL cells through the NF-κB pathway downstream a wide variety of signal transducers, including PLCG2, SYK or LYN, among others [
248].
The second-most frequent mutations found in CLL patients who fail on ibrutinib treatment are
PLCG2 mutations [
249]. The
PLCG2 gene encodes Cγ2, the protein immediately downstream of BTK, and its mutations mostly have an activating effect, resulting in continuous BCR signaling independent of the BTK activity [
243,
249].
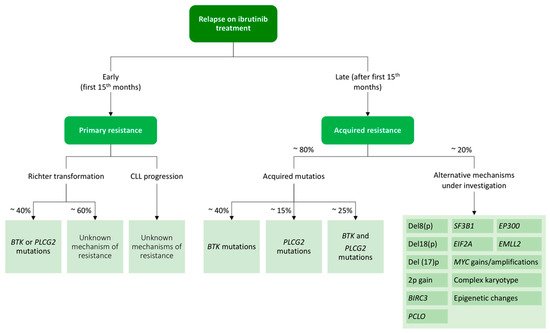
The acquired mutations in these genes have been detected in 80% of patients with ibrutinib failure and CLL progression. Resistance usually develops between the second and fourth year of ibrutinib treatment, but
BTK and
PLCG2 mutations might be detected at low allelic frequencies up to 9–15 months before CLL progression [
115,
244]. In contrast to CLL progression on ibrutinib, which tends to occur later in therapy (after 12 months of attaining a response), Richter transformations mostly occur during the first 1 to 2 years of treatment [
250,
251].
On the other hand, there are approximately 20% of patients in whom
BTK and
PLCG2 mutations cannot be identified. For them, alternative mechanisms of resistance such as 8p deletion or additional driver mutations have been described and are shown in [
252].
Figure 4. Resistance mechanisms to ibrutinib treatment in chronic lymphocytic leukemia.
Intrinsic resistance to ibrutinib is extremely rare and, conversely, has not been studied in depth. Three independent studies analyzed pretreatment samples from patients who relapsed on ibrutinib treatment and failed to find mutations at that moment. In patients that relapse early (first fifteen months), it is necessary to rule out a transformation into a high-grade lymphoma [
242,
253].
Different strategies have been suggested to overcome ibrutinib resistance, highlighting the use of TA targeting other pathways such as PI3K or BCL-2 or the use of reversible BTK inhibitors [
253].
2.3.2. Venetoclax Resistance
The resistance mechanisms of venetoclax are not as well-defined as those occurring after ibrutinib failure. This could be due not only to the later development of the drug but, also, to the implication of different independent molecular mechanisms. Similar to what happens with BTK mutations, a mutation at the G101V in
BCL2 has been implicated in the reduction of venetoclax binding to BCL2. This mutation was found in almost half of patients that progressed under venetoclax in a recent study of a small cohort of cases in 2019. Another mutation in BCL2, D103Y, has been also associated with venetoclax [
254]. This and other mutations could coexist in same patients but as independent clones with different growth dynamics [
254,
255].
BCL2 mutations were identified several months prior to clinical relapse (~25) [
256] ().
Besides
BCL2 point mutations, other candidate resistance-associated aberrations have been reported, including mutations in the antiproliferative
BTG1 gene, aberrations of
CDKN2A/B, the overexpression of
MCL1 and
BCL-XL (pro-survival proteins) and the amplification of
AMP-1, which can affect the OXPHOS pathway in mitochondria [
155,
256,
257].
2.4. COVID-19 and CLL Treatment
The COVID-19 pandemic complicates the current clinical practice for CLL patients, making it more challenging. CLL patients are a population particularly susceptible to SARS-CoV-2 infection, with a high fatality rate (~32–34%) [
258,
259,
260]. This is not surprisingly, as many of these patients harbor high-risk factors for developing severe COVID-19 (age, comorbidities and immunodeficiency) [
261].
A recent study noted that ibrutinib may have a lung-protective effect and may attenuate inflammatory responses due to its inhibitory tyrosine kinase mechanism of action [
262]. Thus, there is much debate on whether patients under BTKi should discontinue treatment if they contract the virus. Evidence is controversial and comes from case reports and a European retrospective study in which patients treated with ibrutinib had a better hospitalization rate [
259,
263,
264]. On the other hand, an American retrospective study did not find this protective effective, even though most cases discontinued BTKi treatment after being infected with SARS-CoV-2. In addition, the second-generation BTKi acalabrutinib was used in a retrospective cohort of 19 severe COVID-19 patients without CLL, with encouraging results [
265]. Hopefully, ongoing prospective clinical trials will clarify if targeting inflammation with a BTKi is a good strategy for COVID-19. In the meanwhile, expert recommendations advocate to limit the patient´s exposure to potential nosocomial SARS-CoV-2 and hold therapy until after recovery of the infection [
266]. If the decision is to continue treatment with BTKi, special care must be taken towards the medical interactions and the hemorrhagic risk, as most critical patients are under an anticoagulant treatment in this phase of the disease.
 +1 point
+1 point