1000/1000
Hot
Most Recent
 +1 point
+1 point
Protein solubility is based on the compatibility of the specific protein surface with the polar aquatic environment. The exposure of polar residues to the protein surface promotes the protein’s solubility in the polar environment. The application of 3D GAuss function allows identification of accordant/discordant regions in proteis. The discordant ones usually represent the localisation of biological activity.
There are two environments of biological activity for proteins: an aquatic environment, and a membrane environment. Hence, the possibility of protein solubility is critical to protein activity. Experimental analysis of the solubility of proteins is supported by theoretical models, which is why many methods and computational tools aimed at determining the degree of solubility are available in the network [1][2]. However, the experimental techniques deliver the important basic information, including the influence of mutations [3][4][5]. The environment other than the water in the cell is the membrane environment, composed of amphiphilic molecules. The proteins anchored in the membrane can reveal the sensitivity of the polypeptide chain to its surroundings [6]. The aggregation of proteins can be treated as partial non-solubility, which is sometimes necessary in order to ensure biological activity, as well as loss of activity, as is observed in the misfolded proteins [7][8][9][10][11][12][13][14][15][16][17][18]—of which the amyloids are spectacular examples [19]. Recently, the intensively applied machine learning technique (applied in PROSO, for example) enables the prediction of protein solubility in heterologous expression in Escherica coli [20][21].
In the present study, a model called the fuzzy oil drop (FOD) [22] was used to reveal the differentiation of solubility and predisposition to the formation of the fourth-order structure, as well as the interaction of the protein with the cell membrane. The arrangement of the hydrophobicity in accordance with the proposed model justifies the high solubility of downhill- and fast-folding proteins [22].
The FOD model is based on the assumption that the idealized hydrophobicity distribution for a fully soluble system of bipolar molecules is the distribution expressed by the 3D Gaussian function, reflecting the hydrophobicity distribution in the spherical micelle. The values of the function spanned on the protein (sigma parameters adjusted to the size of the molecule) express the expected level of hydrophobicity at a given point. Each deviation—whether local, or covering the entire molecule of a protein or complex—identified on the basis of differences between the idealized distribution and that observed in a given protein—assesses its degree of maladjustment to the assumed distribution. The type of mismatch—local exposure of hydrophobic residues and/or local hydrophobic deficit—is interpreted either as a potential possibility of complexing another protein [23], or as the potential possibility of ligand complexation—often related to a function or even a substrate, as is the case with enzymes [24].
Type III antifreeze proteins are proteins whose biological activity is related to their solubility. Their presence prevents water from taking the structural form of ice [25][26]. This work does not concern the analysis of antifreeze proteins (although they are represented in large numbers here). Their presence is related to their high solubility. These proteins are examples of proteins showing high compatibility of the O with the T distribution. This means that the surface of the protein is covered with polar groups which, by imposing a structuring of the surrounding water, prevent it from taking the structural form of ice. This imposition of the structuring of water molecules appears to be a mechanism that prevents water from freezing in organisms producing antifreeze proteins [27]. Proteins from this group were selected from short chains of monomeric form, through longer chains containing domains up to the form of a complex. The characteristics of these proteins, given by the parameter RD, are presented in Table 1.
Table 1. RD parameters for representatives of the soluble protein group. This group includes type III antifreeze proteins of various structures: single chains of varying lengths, a two-domain protein, and a dimer.
| PDB ID | Characteristics | RD—Complete Chain | Selected Fragments | RD |
|---|---|---|---|---|
| 4UR4 | 66 aa | 0.289 | ||
| 6JK4 | 126 aa | 0.489 | ||
| 4UR6 | 64 aa—dimer | 0.638 | Chain A Chain B |
0.298 0.280 |
| 1C8A | 134 aa—two domains | 0.659 | Dom1 Dom2 |
0.293 0.280 |
Type II and III antifreeze proteins represent structures with very low RD values. Here, an example of this group of proteins is a 66-amino-acid chain in a fish antifreeze protein from zoarces viviparus—zvafp13 (PDB ID 4UR4). The O distribution in this protein is very close to that of the T distribution, which guarantees the high solubility of this protein. It should be noted that the compatibility of the O distribution with the T distribution indicates the presence of a centric concentration of hydrophobicity with the presence of the polar surface of the protein. Similarly, the double-length protein—the Ca2+-dependent type II antifreeze protein (PDB ID 6JK4)—also shows a distribution matching the idealized one. This agreement indicates exposure of polar residues to the surface.
The dimeric structural form of type III fish antifreeze protein from zoarces viviparus—zvafp6 (PDB ID 4UR6)—deposited in the PDB does not appear to be the form found in nature. The monomer status shows very low RD values, which means full surface coverage by polar residues. The globular (close to spherical) structure of each chain of this protein is accompanied by very little intermolecular contact. Intermolecular contact relies mainly on several interactions, based on the interactions of polar groups. The impact of the electrostatic type in the aquatic environment, in the form of limited contact, is highly unlikely. At the same time, a very low RD value for single structures indicates the full availability of the surface for interactions with water.
The intramolecular dimer antifreeze protein rd3 (PDB ID 1C8A) has an RD value of >0.5 (Figure 1). The structure of this chain consists of two domains that are almost completely isolated from one another, linked only by the segment 55–72 in the form of a loose loop. This loop likely shows significant structural variation, which may result in the free movement of domains with a clearly polar surface. The imposition of a structure different from that of ice on the surrounding water molecules is apparent here. The high polarity of the protein’s surface guarantees its high solubility, without which its biological function of resisting the structuring of water molecules into the form of ice would be impossible. Figure 2 and Figure 3 show the high compatibility of the O and T distributions in the proteins under discussion.
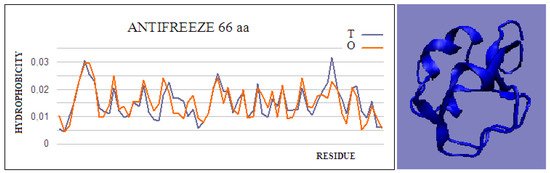
Figure 1. Hydrophobicity profiles. T: idealized (blue); O: observed (red), together with 3D presentation showing the highly globular form of this protein (PDB ID 4UR4). The program VMD was used to present the 3D form http://www.ks.uiuc.edu/Research/vmd/, accessed on 15 March 2021.
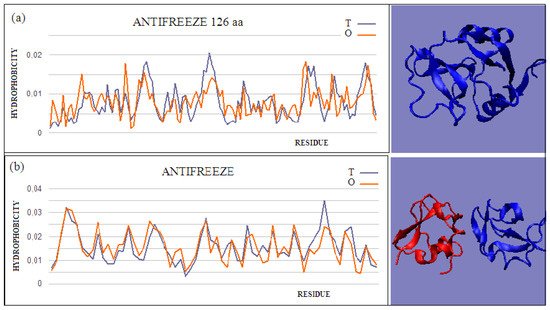
Figure 2. Two examples of proteins with high T and O compatibility of longer chains: (a) PDB ID 6JK4, and complex form (b) PDB ID 4UR6, together with 3D presentation, respectively. The program VMD was used to present the 3D form http://www.ks.uiuc.edu/Research/vmd/, accessed on 15 March 2021.
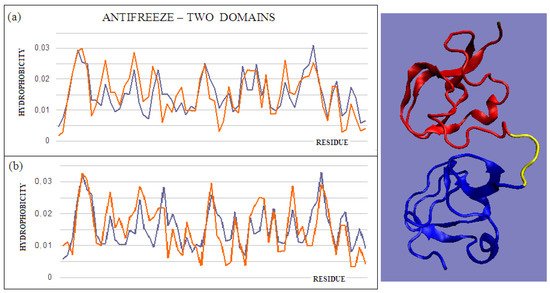
Figure 3. T and O profiles of an antifreeze protein with a two-domain structure. (a) N-terminal domain, and (b) C-terminal domain with a 3D presentation of this protein. Color-distinguished domains—yellow fragment: linker with high structural freedom (PDB ID 1C8A). The program VMD was used to present the 3D form http://www.ks.uiuc.edu/Research/vmd/, accessed on 15 March 2021.
The domain-swapping phenomenon is the exchange of structural elements between proteins, resulting in the formation of dimeric (or polymeric) forms. A chain fragment of one protein interacts with another protein in order to complete its super-secondary structure. This fragment, leaving its position in the native protein, opens the site for complexation of a fragment of the chain of another protein. The reaction can cause a domino effect. The domain-swapping phenomenon is observed relatively often [28][29][30]. The answer to the question about the mechanism of this phenomenon, based on the fuzzy oil drop model, shows that the structures of the hydrophobic core complement one another. Two sample proteins will be discussed using the fuzzy oil drop model. One is limited to docking a short chain that conforms to a slightly deformed beta-barrel or beta-sandwich domain arrangement (PDB ID 2CO2) (Figure 4). In the second case, the replacement of chain sections concerns the chain’s short fragments. The structure of the domain Salmonella enterica SafA pilin in complex with a 19-residue SafA Nte peptide shows the status of the idealized distribution. This means that it can function as an independent structural unit in the aquatic environment. Complexation of the 16-amino-acid polypeptide chain, however, results in an additional reduction in the RD value, which means that the status of the complex is more favorable for the structure as a whole.
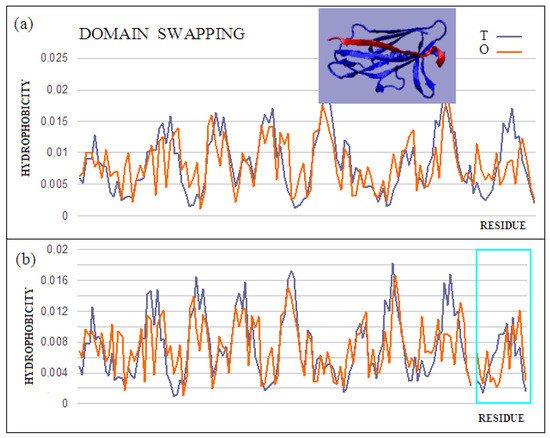
Figure 4. Characteristics of the domain-swapping complex present in an outer membrane protein (PDB ID 2CO2). (a) T and O profiles for chain A, and (b) T and O profiles for chain A with chain B incorporated. Turquoise frame: chain B; Inset: 3D structure; –red: chain B. The program VMD was used to present the 3D form http://www.ks.uiuc.edu/Research/vmd/, accessed on 15 March 2021.
This is also evidenced by the change in the status of the system itself: a slightly deformed beta-barrel shows an RD of 0.428, and in the composition extended by the presence of an additional polypeptide shows an RD of 0.416. Similarly, the status of the adjacent segments to the incorporated peptide shows an RD of 0.664, while the status of the same system in the presence of a polypeptide decreases to an RD of 0.533. The presence of the polypeptide in each of the considered systems turns out to be advantageous in reducing the RD value, which means obtaining a distribution close to the idealized distribution. This, in turn, favors obtaining a micelle-like structure, which is a favorable system in the water environment for bi-polar molecules, with different degrees of differentiation of the polar to the hydrophobic parts. Thus, a micelle-like structure is the optimal solution here. The status of the components of this complex is shown in Table 2.
Table 2. A set of values of the RD parameter for structures created via domain swapping.
| PDB ID | Target Mol. | RD | Incorporated | RD | Complex | RD |
|---|---|---|---|---|---|---|
| 2CO2 | Pilin domain, 48–170 | 0.472 | N-terminal 27-45 |
0.725 | Complex | 0.456 |
| Beta-barrel | 0.428 | Beta-barrel with beta-fragment incorporated | 0.416 | |||
| 3CRF | Chain A | 0.509 | Chain C | 0.731 | Complex A+C | 0.500 |
| 2J6R | Chain A | 0.669 | Chain A(no 24-31) with B(24-31) |
0.662 |
The core pilin domain (PDB ID 3CRF) structure is more complex. It consists of a system of two chains (A, B) and a C peptide. The B domain provides fragments 0–19 of the A domain. However, the B domain also includes the C 0–19 peptide. The structure of the beta-structural system forms a deformed beta-barrel. Fragments 0–19 are part of these deformed beta-barrels. The complex does not exhibit an ordered structure of the fuzzy oil drop model. This is mainly due to the absence of a common hydrophobic core. In a central part of the complex, where the hydrophobic core is expected, the loosely packed interface is present. Chains A and B, treated as individual structural units, show a status minimally exceeding the adopted threshold level (RD = 0.5). Connecting fragments 1–19 results in the reduction of the RD value to 0.500 (Table 2). In the given examples, the presence of an incorporated chain fragment from another protein molecule results in a lower RD value, which means a better adjustment to an idealized state.





