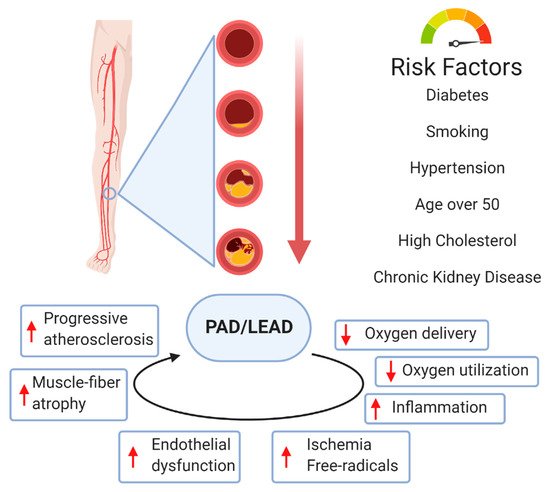
LEAD usually involves atherosclerotic disease in the abdominal aorta, iliac, and femoral arteries. The pathophysiology of atherosclerotic plaque involves complex interactions between cholesterol metabolism and vascular cell activity. It is also known that disturbance of the laminar arterial flow in PAD plays an essential role in the adhesion of inflammatory blood cells to the arterial wall and plays a role in plaque generation
[14]. The hemodynamic consequences of atherosclerotic plaque depend on the degree of stenosis/occlusion. A narrowed vessel progresses towards chronic total occlusion. LEAD patients demonstrate high grades of inflammation and active oxidative stress, which are vital mechanisms in PAD pathophysiology
[14]. There is an interest in the vaso-protective effects of heme oxygenase 1 (HO-1) as a potential antioxidant by affecting the proliferation, migration, and adhesion of smooth vascular muscle cells, endothelial cells, and leukocytes
[15]. It was previously suggested that PE acts on endothelial functions by regulating genes involved in modulating oxidative metabolism, cell apoptosis, cell growth and proliferation, and endothelial vascular nitric oxide synthase (eNOS)
[14][16].
Vessel wall remodelling and angiogenesis in peripheral arteries appear to be the most important tissue response process to the atherosclerotic injuries
[17]. Extracellular matrix (ECM) provides a mechanical scaffold and support to cell migration regulated by cytokines, growth factors, and enzymes, such as matrix metalloproteinases (MMPs)
[18][19]. Hernandez-Aguilera et al. suggested that atherosclerosis of the lower extremities is due to an excessive reparative response, which includes factors favoring ECM degradation
[19][20]. Decreased levels of structural proteins of the arterial wall are highly influenced by MMP activity
[19]. These proteins also have defined roles in maintaining normal function and migration of smooth muscle cells
[21]. Connective tissue turnover in the structural and signaling properties of the arterial cells plays a central role in the LEAD development
[22]. Single nucleotide polymorphisms of the genes encoding for some MMPs have also been associated with the risk of developing LEAD. Blankenberg et al.
[23] reported a study that focused on two MMP-9 gene polymorphisms: the MMP-9/C-1562T promoter polymorphism, which influences the transcriptional activity of the MMP-9 gene, and the exonic MMP-9/R279Q polymorphism, which leads to an amino acid exchange in the catalytic domain of MMP-9. The authors described a significant association between the R279Q polymorphism and cardiovascular events in patients with stable angina.
Arterial calcification was previously considered a passive degenerative process but is now recognized as a complex process actively regulated by several cell molecules
[24]. Arterial calcification mainly involves the intima and media, and is associated with cardiovascular risk factors, such as diabetes, hypertension, hyperlipidemia, and chronic kidney disease
[25]. Arterial calcification is related to the apoptosis of vascular smooth muscle cells and macrophages
[26]. Studies provided by Hui laboratory
[27] focused on the genetic variants of the matrix Gla protein (MGP), a protein known as a key player in in vivo inhibition of calcification. The authors analyzed rs4236, rs1800801, and rs1800802 variants of the
MGP gene, and showed association with calcification on the arterial wall but not with calcification in atherosclerotic plaques
[27]. Other studies showed that different types of arterial calcification develop through various molecular mechanisms in different vessel types
[28]. In contrast to carotid and coronary arteries, arterial calcification of LEAD is mostly located in the media.
Several genotypes serve as potential risk factors for atherosclerosis. However, evidence of their clinical relevance is weak. Some of the most common risk factors for PAD (diabetes, dyslipidemia) are heritable. However, PAD may also result from genetic factors acting independently. Identification of such genes may provide insights into pathophysiologic mechanisms of PAD progress and facilitate the development of novel therapeutic approaches
[29]. In contrast to coronary artery disease, relatively few genetic variants that influence susceptibility to PAD have been discovered because there may be more significant clinical and genetic heterogeneity in PAD patients. Definitively, genetic factors may have an impact on the early onset of PAD in young adults, including these mechanisms affecting the process of inflammation, thrombosis, and the metabolism of cholesterol and homocysteine. Among these genetic disorders, familial hypercholesterolemia or hyperhomocysteinemia are the most known. For example, Flex et al.
[30] explored the association between the interleukin-6 gene (IL-6)-174 G/C single nucleotide polymorphism (SNP) and the risk of peripheral artery occlusive disease. The authors concluded that the analyzed polymorphism is important in the pathophysiology of ischemic diseases of the lower extremity
[30]. Several other candidate genes involved in the process of atherosclerosis and regulating inflammation and coagulation pathways and vascular matrix regulation were also analyzed, such as β-fibrinogen, eNOS, MTHFR, and glutathione S-transferase
[31]. However, any reported associations between variants in these genes and PAD have not been confirmed
[29].
3.3. Angiogenesis
Angiogenesis is the process of new capillary formation from pre-existing capillary beds that involves proliferation, sprouting, and migration of endothelial cell migration. Angiogenesis occurs naturally during wound healing, tissue growth, and repair. The process of angiogenesis is highly controlled, dependent on a balance of both pro-angiogenic and anti-angiogenic factors. The process of angiogenesis results from complex interactions between growth factors, endothelial cells, pericytes, fibroblasts, smooth muscle cells, and the extracellular matrix. As a result of these interactions, extracellular proteolysis, endothelial cell migration, proliferation, and differentiation, and finally, vascular wall remodeling can occur
[4]. Vascular endothelial growth factor (VEGF) is the main factor stimulating angiogenesis in response to tissue hypoxia
[36]. Angiogenesis is primarily stimulated by tissue hypoxia via activation of hypoxia inducible factor (HIF). Specifically, in the absence of cellular oxygen, hypoxia-inducible factors (HIFs) are activated and translocated to the nucleus where they upregulate genes including VEGFA, angiopoietins, and nitric oxide
[1][2][37]. Through signaling by VEGF, mature endothelial cells (ECs) are directed to sites of hypoxia to participate in the formation of new blood vessels. The endothelial cell activation is associated with cytokine release, initiation of vasodilation, and increased endothelial cell permeability. VEGF promotes the release of many proteolytic factors, such as matrix metalloproteases, which degrade the extracellular matrix and facilitate endothelial cell migration
[38]. Once a functional vascular network is formed, the new vessels are remodeled to become a mature vessel system
[32]. Angiogenesis describes the vascular tissue remodeling and is characterized by the expansion of existing capillary density to reach ischemic sites, maintaining the lower extremity function in patients with LEAD.
3.4. Arteriogenesis
As a distinct process from angiogenesis, arteriogenesis refers to the growth of new arteries and arterioles either de novo or from pre-existing arterial collaterals
[3][39]. It mainly involves the proliferation of vascular endothelial cells (ECs) and smooth muscle cells (SMCs)
[40]. As a result of the occlusion of the main arterial trunk, the pre-existing arterio-arteriolar anastomoses between interconnected perfusion territories can undergo adaptive enlargement that develops into a functional network of collateral arteries
[32]. Arteriogenesis is critical to the restoration of tissue perfusion following the development of a functionally significant decrease of arterial inflow during LEAD
[41]. The transformation of native microvascular collateral arterioles into functional arteries with consequent recovery of blood flow is particularly observed after ischemic injury
[42].
It was previously suggested that vessel segments could adapt to the amount of flow
[43]. Collateral growth is driven by hemodynamic forces and leads to initial vasodilation due to increased levels of nitric oxide
[44]. While hypoxia is the primary driver of angiogenesis, arteriogenesis is mainly induced by a combination of shear stress and other mechanical factors
[36][39][41]. Pulsatile shear stress activates the cascade of events that leads to the development of collateral circulation
[4][45]. Several genes are controlled by shear stress-responsive elements in their promotor, and fluid shear stress influences these genes’ expression. The role of mechano-sensors and transducers that convey the shear stress message during collateral remodeling has been suggested as a mechanism directing neovascularization
[46][47][48]. A fluid shear stress-associated transient receptor potential cation channel, subfamily V, member 4 (trpv4), turned out to be upregulated transiently after endurance training
[49][50]. Shear stress increases the expression of an isoform of connexin, connexin-37, in endothelial cells
[51].
Arteriogenesis consist of two phases: the early inflammation phase and the later phase of vessel diameter increase, remodeling, and maturation. Hemodynamic forces in the collateral vessels are the primary stimulus to initiate arteriogenesis. As a result of the increased shear stress force, endothelial cells express monocyte adhesion molecules: platelet endothelial cell adhesion molecule (PECAM-1), MCP-1, intracellular adhesion molecule (ICAM-1), and vascular cell adhesion molecule (VCAM-1)
[52]. Endothelial cells regulate adhesion molecule gene expression through a mechano-transduction process by specific shear stress receptors
[32]. Cytokines and cell adhesion molecules attract monocytes to adhere and invade the vascular wall. Recruited monocytes infiltrate the vessel wall and transform into macrophages. Once activated, they produce TNF-α and attract more monocytes. Recruitment of circulating monocytes and resident macrophages promotes arteriogenesis by their ability to secrete metalloproteinases, chemokines, and growth factors
[53]. The proliferation of the endothelium is followed by smooth muscle proliferation and migration to form a new neointima. Little is known about the mechanisms triggering SMC proliferation in arteriogenesis. Growth factors, such as fibroblast growth factor 2 (FGF-2) and platelet-derived growth factor-BB (PDGF-BB), are essential for SMC proliferation in arteriogenesis
[54]. Finally, enlargement of the blood vessel occurs by remodeling the adventitia through fibroblast activation and proliferation. These events lead to new collateral vessel development in postnatal life.
Arteriogenesis differs from angiogenesis in several aspects, the most important being the dependence of angiogenesis on hypoxia and the dependence of arteriogenesis on inflammation. Arteriogenesis occurs in tissues near arterial stenosis/occlusion, whereas peripheral ischemic regions undergo angiogenesis, which is the growth of new capillaries
[55]. Collateral vessels resulting from arteriogenesis are typically surrounded by normoxic tissue
[56]. While molecular regulation of angiogenesis is well analyzed, events regulating arteriogenesis are still controversial. As well as for angiogenesis, VEGF is also critical to arteriogenesis
[57]. In arteriogenesis, VEGF activation of extracellular signal-regulating kinase 1/2 (ERK1/2) induces endothelial cell proliferation, network formation, and increased lumen size. Disruption of VEGF-induced endothelial ERK1/2 signaling results in decreased arteriogenesis
[58]. The activation of this signaling is modulated by NRP-1
[59]. VEGF, TGF-α, and FGF-2 stimulate the proliferation of endothelial cells, whereas TGF-β, GM-CSF, monocyte chemoattractant protein-1 (MCP-1), and PDGF stimulate arteriogenesis through the proliferation of smooth muscle cells
[60]. The proliferation of endothelial cells and smooth muscle cells leads to lumen size expansion of the collateral artery
[61]. In contrast to angiogenesis, arteriogenesis requires a coordinated response that involves multiple cell types, not only endothelial and vascular smooth muscle cells
[41]. Several inflammatory cells, including lymphocytes, natural killer (NK) cells, macrophages, and mast cells, were also suggested to play a role in arteriogenesis
[62][63][64]. The presence of inflammatory cells is critical as these cells serve as the major source of VEGF in the absence of tissue ischemia
[41][65][66]. Mast cells have also been associated with arteriogenesis and collateral formation
[67][68]. The proinflammatory response during arteriogenesis is beneficial in restoring blood flow, but may lead to enhanced progression of atherosclerosis
[68].
Collateral circulation occurs between arterioles in most healthy tissues and in pathological conditions like ischemic injury
[39]. Arteriogenesis can be induced by regular exercise or muscle loading in humans in physiological conditions
[33][49][69]. A more regular exercise regimen was also proposed to be required for sufficient arteriogenesis to compensate for an arterial occlusion in LEAD
[49]. It explains why significant stenosis/occlusion of main arteries may remain asymptomatic in patients with chronic LEAD for many years. Exercise therapy can influence the vascular system supply to the ischemic limb by promoting both angiogenesis and arteriogenesis
[33]. An increase of angiogenic and arteriogenic factors, such as VEGF and HIF-1 alpha, was observed after regular exercise
[70]. Additionally, higher concentrations of heat shock proteins and the enzyme nitric oxide synthase (NOS) in blood during exercise therapy was noticed
[71]. Nitric oxide (NO) is an important cellular signaling molecule produced in high levels in muscle by neuronal NOS
[33]. Under ischemic conditions, the role of NO in vasodilatation is increased compared with non-ischemic conditions, which results in an up-regulation of endothelial NOS (eNOS)
[72]. Regular exercise training in the patient with LEAD leads to similar mechanisms of vascular adaptation such as increased fluid shear stress and promotes arteriogenesis. After successful revascularization, collateral arteries shrink or disappear as the main blood flow is directed back through the re-opened artery or vascular reconstruction. Supervised exercise therapy is considered to be a potential therapeutic option in chronic LEAD patients under conservative treatment.
 +1 point
+1 point