1000/1000
Hot
Most Recent
 +1 point
+1 point
Glutathione (GSH) is the most abundant non-protein thiol, and plays crucial roles in the antioxidant defense system and the maintenance of redox homeostasis in neurons.
In 1888, de Rey-Pailhade described a substance with the property of reducing sulfur to hydrogen sulfide in extracts from yeast; he named this molecule ‘philothion’ [1], meaning “love of sulfur” in Greek [2][3]. Heffter suggested that cysteine (Cys) was involved in this molecule [4], although the structure of this substance was not revealed until the 1920s. Since that time, many researchers have been engaged in the extraction and synthesis of glutathione (GSH), including three Nobel laureates. In 1921, Frederick G. Hopkins isolated a dipeptide containing glutamate (Glu) and Cys from yeast and animal tissues, and named it ’glutathione’ [5]. However, de Rey-Pailhade thought that glutathione was a side chain of philothion, which was speculated to be a protein [3]. Other researchers isolated GSH from yeast, blood, and liver, and suggested that GSH was not a simple dipeptide composed of Glu and Cys [6]. Ultimately, in 1929, Hopkins redetermined that GSH was a tripeptide containing Glu, Cys, and glycine (Gly) [7]. In the same year, Hopkins was awarded the Nobel Prize in Physiology or Medicine for his work on vitamins, not GSH. Also in 1929, Edward C. Kendall crystallized GSH and identified its chemical structure [8]; Kendall went on to be awarded the Nobel Prize in Physiology or Medicine for his work on corticosteroids in 1950. Finally, Vincent du Vigneaud first reported the synthesis of GSH in 1936 [9]; he was awarded the Nobel Prize in Chemistry in 1955 for his work on biochemically important sulfur compounds, especially for the first synthesis of oxytocin. As evident from this thumbnail history, GSH inspired the interest of some of the most prominent researchers of the early twentieth century. More than 130 years after its discovery, GSH is still a promising therapeutic target for the treatment of neurodegenerative diseases.
GSH is a major antioxidant that maintains the homeostasis of redox states in cells, and plays important roles in maintaining the physiological functions of all cells in vivo. The thiol (sulfhydryl, SH) residues play an important role in maintaining the redox state homeostasis intracellularly. In mammals, Cys and methionine (Met) are particularly important as thiol-containing amino acids [10], but GSH is the most abundant thiol-containing substance (derived from a non-protein) in all kinds of cells. The functions of GSH in living cells are diverse, and include roles in maintenance of the intracellular antioxidant system, redox balance, Cys transport/storage, cell signaling, regulation of some enzyme activities, gene expressions, and cell differentiation/proliferation [11]. GSH is especially abundant in the liver and kidney [12], both of which utilize the transsulfuration pathway to produce Cys from Met via homocysteine [13], while it is present at lower levels in the brain, where the regulatory system for GSH synthesis is independent of that in peripheral tissues. Therefore, the molecular mechanisms underlying GSH dysfunction in the brain differ from those in peripheral tissues.
All of the major biological processes of GSH involve the redox state of the thiol residue within the GSH molecule. Two molecules of GSH are oxidized to produce one molecule of GSH disulfide (GSSG) in order to eliminate reactive oxygen species (ROS)/reactive nitrogen species (RNS), or to maintain intracellular redox homeostasis, and GSSG can be reduced back to two GSH molecules via reaction with GSH reductase (GR). The intracellular GSH/GSSG ratio is 100 or more in the steady state, but decreases to 10 or less under oxidative stress conditions [14]. Proteins oxidized by ROS/RNS are reduced by glutaredoxins (Grxs) or thioredoxins (Trxs) [15] (Figure 1). Functionally, Grxs and Trxs share many common features; however, Grxs are more versatile than Trxs in terms of their substrate selectivity and reaction mechanisms [16]. The isoforms of Grxs and Trxs are known as cytosolic Grx1 and Trx1, and as mitochondrial Grx2 and Trx2 in mammals [16][17]. These isoforms are involved in controlling intracellular redox signaling for the cellular processes of apoptosis and proliferation [17][18]. Grx2 and Trx1 are also found in the nucleus. Many transcription factors are known to undergo reduction by Grxs and Trxs. In order for a transcription factor to bind to DNA, the thiol groups of Cys residues in the DNA-binding site should be in the reduced form. The thiol groups mainly exist in their oxidized forms, and are reduced in the process of becoming activated to enable DNA binding. Subsequently, the oxidized Grxs and Trxs are reduced back via reaction with GSH and Trx reductase (TrxR), respectively. GSH functions as an enzyme cofactor for Grxs, which are low-molecular-weight redox enzymes that are also known as thiol transferase, to maintain cellular redox homeostasis, and also acts as the primary reductant of the disulfide bonds of oxidized proteins [15] (Figure 1). However, excessive oxidative stress causes irreversible oxidation of the thiol residues and impairs cellular protein function [19][20]. In particular, Cys residues in active sites or functional motifs of intracellular proteins are important for their protein functions. Oxidative stress by ROS/RNS on the Cys residues in proteins can cause irreversible modifications that lead to critical dysfunction of the proteins [21]. GSH can also react with intracellular protein thiol residues to protect protein functions related to enzyme activities, DNA binding by transcription factors, and protein stability [22][23][24]. Therefore, under such oxidative stress conditions, GSH reversibly binds to the thiol residues to prevent irreversible changes in proteins due to oxidative stress. This post-translational modification, called “glutathionylation”, is reversible and protects the intracellular signal transduction system against oxidative stress [19] (Figure 1). The caspase family of Cys proteases, which induces cell apoptosis, would be a potential target for glutathionylation. Caspase-3, an important regulator of apoptotic responses, can undergo glutathionylation, leading to the inactivation of caspase-3 by GSSG in a dose- and time-dependent manner [25], suggesting that apoptosis can be regulated by glutathionylation. Once the cellular environment is free from oxidative stress, the disulfide bonds in the proteins are reduced back by Grx to function normally under physiological conditions [15]. Thus, the regulation of the redox state by intracellular GSH is extremely important for maintaining cellular functions under both physiological and pathological conditions. Especially in the brain, the regulatory mechanism of GSH function is more fragile in neurons than in glial cells, and intracellular GSH levels are also lower in neurons than in glial cells [26]. Under some pathological conditions, decreased GSH levels could have critical influences on neuronal activities, leading to neurodegeneration.
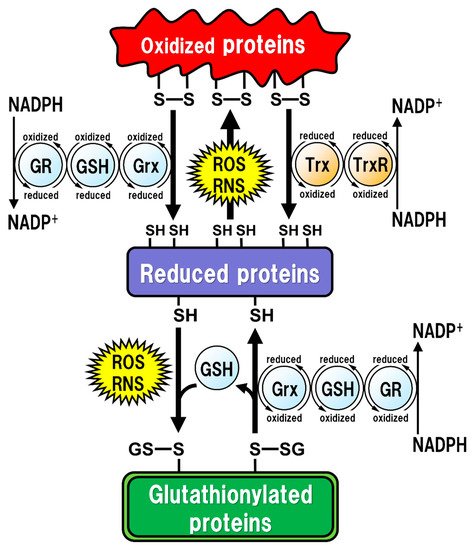
Figure 1. Regulation of the intracellular protein redox state by glutathione (GSH), glutaredoxin (Grx), and thioredoxin (Trx). Reactive oxygen species (ROS) and reactive nitrogen species (RNS) cause protein dysfunction, which is induced by the oxidation of thiol (SH) residues to form disulfide (S-S) bonds in the active site. Grx and Trx regulate protein function by reducing the S-S bonds of the substrate proteins. Consequently, Grx and Trx themselves result in the oxidized forms, which are reduced back by GSH and Trx reductase (TrxR), respectively. Oxidized GSH (GSSG) is reduced back to GSH by GSH reductase (GR). Both oxidized TrxR and GR are reduced by receiving electrons from nicotinamide adenine dinucleotide phosphate (NADPH). Under oxidative stress conditions, GSH can bind to cysteine residues (GS-S) in a process known as ‘S-glutathionylation’ to prevent the irreversible dysfunction of the proteins. Grx also functions in the deglutathionylation of the GS-S containing proteins to resume protein functions under physiological conditions.
GSH occupies approximately 95% of non-protein thiol groups in vivo and is ubiquitously present in mammalian cells at concentrations of 0.5 to 10 mM, depending on the tissues [27][28]; these concentrations are 10 to 100 times higher than the concentrations of Cys in mammalian cells [29]. GSH is the major intracellular thiol compound, and is made from Glu, Cys, and Gly by two-step enzymatic reactions requiring ATP. The first step of these reactions is mediated by Glu-Cys ligase (GCL), and the second by GSH synthetase (GSS). In intracellular GSH synthesis, GCL can be the rate-limiting enzyme under the condition that all substrates are sufficiently present for the reactions, but the intracellular Cys concentration is much lower than those of Glu or Gly under physiological conditions, suggesting that the Cys availability is limiting for GSH synthesis [30][31]. GCL is comprised of both a catalytic (GCLc) and a modulatory (GCLm) subunit. GCLc is responsible for all of the enzyme activity of GCL, which is regulated via feedback inhibition by GSH [32]. Most GSH is present in the cytoplasm, where it is synthesized in mammalian cells [33]. Although mitochondria contain about 5–15% of all the GSH in the cell [34], they cannot synthesize GSH by themselves because they lack GCL [33]. The finding that GCLc- or GSS-deficient mice are non-viable in the embryonic period [35][36], while GCLm-deficient mice are viable and fertile with decreased GSH levels in the tissues compared to those of wild-type mice [37], suggest that GSH is essential for embryogenesis.
The highest GSH concentration in the body is in the liver (about 5 to 10 mM) [12], but hepatocytes can also produce Cys for GSH synthesis from Met via the transsulfuration pathway [38]. GSH in the liver is then released systemically, but it is decomposed in the blood, with the result that the blood GSH concentrations (approximately 2 to 20 µM) are hundreds to thousands of times lower than those in the liver [28]. In addition, GSH cannot directly enter the brain due to the existence of the blood-brain barrier (BBB). Moreover, extracellular GSH cannot be directly transported into the cells, and thus the three amino acids used as substrates for GSH synthesis should be taken up into the cells via transporters.
The brain tissue is generally rich in unsaturated fatty acids, which are targets of oxidative stress, and has relatively low levels of antioxidants or antioxidant enzymes. ROS, such as singlet oxygen (1O2), superoxide (O2·−), and hydroxyl radicals (·OH), are endogenously produced by mitochondria, cytochrome P450 metabolism, peroxisomes, and inflammatory cell activation. Mitochondria generate most ROS, including O2·−, into the matrix and the intermembrane space via the electron transport chain. The steady-state concentration of O2·− is about 5–10 times higher in the mitochondrial matrix than in the cytoplasm or nucleus [39], but the mitochondrial matrix contains Mn-superoxide dismutase (SOD), which can react with O2·− to form hydrogen peroxide (H2O2) (Figure 2). In addition, O2·− leaked into the cytoplasm reacts with Cu/Zn-SOD (SOD1) to form H2O2. H2O2 is toxic to eukaryotic cells at concentrations of 0.1 to 1 × 10⁻3 M, but the reaction with catalase or GSH peroxidase (GPx) can decompose H2O2 to oxygen and water. As a result, the concentrations of H2O2 in mitochondria are maintained in the range of 10⁻⁹ to 10⁻⁸ M [15]. Such high concentrations of H2O2 are unlikely to occur under physiological conditions in vivo. However, overproductions of both O2·− and H2O2 can be induced by mitochondrial dysfunction [40]. The increased H2O2 produces ·OH, which possesses the highest reactivity and the strongest oxidizing power among ROS, via the Fenton reaction (Figure 2). In addition, O2·− reacts with nitric oxide (NO) to generate peroxynitrite (ONOOˉ) (Figure 2), which targets DNA, proteins, and lipids, causing DNA damage, dysfunction of enzymes, receptors, transporters, and membrane channels, as well as protein aggregation, mitochondrial dysfunction, and lipid peroxidation [15]. ONOOˉ is produced approximately one million times faster, and can spread approximately 10,000 times farther over cells, than ·OH [41]. ONOOˉ is more globally toxic within tissues than ·OH, whose toxicities are limited to the local area inside the cells [42]. GSH acts protectively against oxidative stress by reacting directly with NO, O2·−, H2O2, ·OH, and ONOOˉ (Figure 2). GSH also acts as an enzyme cofactor for GPx to degrade H2O2 and hydroperoxides (ROOH), and is involved in detoxifying electrophilic xenobiotics via GSH-S-transferase (GST) [43] (Figure 2). From these protective functions, GSH is considered to play an important role not only under physiological conditions but also under pathological conditions induced by oxidative stress in order to maintain the homeostasis of cell functions.
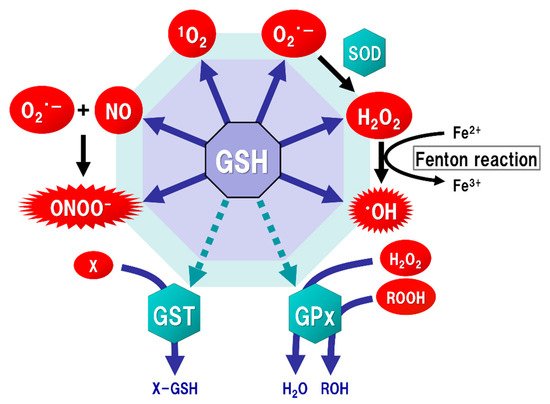
Figure 2. Function of glutathione (GSH) as an antioxidant. Mitochondria generate superoxide (O2·−), which reacts with nitric oxide (NO) to form peroxynitrite (ONOOˉ), a typical reactive nitrogen species (RNS) that is a potent inducer of cell death. O2·− is catalyzed to hydrogen peroxide (H2O2) by the reaction of superoxide with superoxide dismutase (SOD). H2O2 reacts with Fe2+ (Fenton reaction) to form a highly oxidizing radical, hydroxyl radical (·OH). GSH can directly act as an antioxidant (solid arrows) by non-enzymatically reacting with NO, singlet oxygen (1O2), O2·−, H2O2, ·OH, and ONOOˉ. GSH can also indirectly serve as an enzyme cofactor for detoxification (dotted arrows). H2O2 is catalyzed to water and oxygen by GSH peroxidase (GPx), which requires GSH as an electron donor to react with H2O2 and hydroperoxides (ROOH). GSH-S-transferase (GST) can detoxify various xenobiotics (X) via GSH conjugation to excrete toxic compounds from the cell.
In in vitro studies, GSH levels in neurons are lower than those in astrocytes [26], and are increased when the neurons are co-incubated with astrocytes [44]. Neuronal GSH synthesis is supported by astrocytes, which supply GSH precursors to neurons. Notably, neuronal GSH levels in vitro are increased by the administration of Cys, but not Glu, Gly, or cystine, the latter of which is formed by two Cys molecules with a disulfide linkage [44][45]. Both Cys and Met are major sources of mammalian thiols [10], and Cys is an important substrate for GSH synthesis in neurons [46], while astrocytes can utilize both Cys and cystine for their GSH synthesis [45]. The activity of GCL, the rate-limiting enzyme for GSH synthesis, was upregulated in neurons co-cultured with GSH-depleted astrocytes, but the neuronal GSH levels were not increased [47]. These findings suggest that not only neuronal GCL activity, but also the astroglial supply system with Cys-containing precursors, is important in maintaining neuronal GSH levels.
The uptake of Cys into neurons is mainly mediated by excitatory amino acid carrier 1 (EAAC1, in rodents), also known as excitatory amino acid transporter type 3 (EAAT3, in humans) (Figure 3). Five types of EAAT have been reported so far, and their expressions differ depending on the cell type. In the brain, GLAST (also known as EAAT1) and GLT-1 (also known as EAAT2) are primarily distributed in astrocytes, whereas EAAC1 is exclusively expressed in neurons. EAAT4 and EAAT5 are distributed in cerebellar Purkinje cells and neurons of the retina, respectively [48]. All of these transporters can take up extracellular Glu into the cells, but unlike GLAST and GLT-1, EAAC1 can also transport Cys with the same efficiency as Glu [49]. Based on the experimental results using a mutation model of EAAC1, it has been considered that the mechanisms of Glu and Cys uptake by EAAC1 are independent of each other [50]. There were no significant changes in extracellular Glu concentrations in an EAAC1-knockdown animal model [51]. GLAST and GLT-1 act as Glu transporters in glial cells in vivo and are involved in the regulation of Glu concentration in synaptic clefts, whereas EAAC1 is not involved in the regulation of extracellular Glu levels in synaptic clefts, but rather in the regulation of GSH production via extracellular Cys uptake. Moreover, EAAC1-deficient mice exhibit decreased brain GSH levels, vulnerability to oxidative stress in the hippocampus, and age-related learning dysfunction [52]. EAAC1-deficient mice also showed age-dependent loss of dopaminergic neurons in the substantia nigra pars compacta accompanied by increased oxidative stress [53]. EAAC1 is responsible for approximately 70–80% of Cys uptake in neurons [54], and can transport 10- to 20-fold greater amounts of Cys than can GLAST or GLT-1 [49]. Based on these results, the physiological roles of EAAC1 in the central nervous system (CNS) would be involved in the neuroprotective roles mediated by GSH production [55].
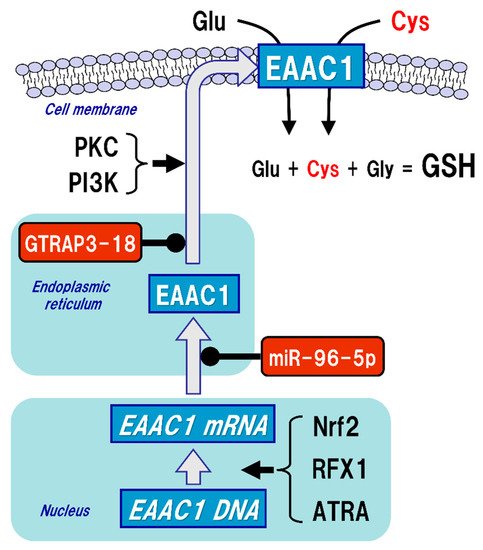
Figure 3. Regulation of excitatory amino acid carrier 1 (EAAC1) expression. Glutathione (GSH) is a tripeptide synthesized from glutamate (Glu), cysteine (Cys), and glycine (Gly). Neuronal GSH synthesis relies on intracellular Cys but not Glu or Gly level. Cys uptake (red font) is subjected to the regulation of both gene expression and post-translational modifications of EAAC1 under facilitative (arrow) and suppressive (black circles) controls. EAAC1 gene expressions are promoted by nuclear factor erythroid 2-related factor 2 (Nrf2), regulatory factor X1 (RFX1), and all-trans-retinoic acid (ATRA). Protein kinase C (PKC) and phosphoinositide 3-kinase (PI3K) activations increase the EAAC1 expression on the plasma membrane. Glu transporter-associated protein 3-18 (GTRAP3-18) and miR-96-5p post-translationally suppress the protein expression of EAAC1, leading to decreased Cys uptake and subsequently decreased GSH synthesis in neurons.





